FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
FORTEO is indicated:
- For the treatment of postmenopausal women with osteoporosis at high risk for fracture (defined herein as having a history of osteoporotic fracture or multiple risk factors for fracture) or who have failed or are intolerant to other available osteoporosis therapy. In postmenopausal women with osteoporosis, FORTEO reduces the risk of vertebral and nonvertebral fractures.
- To increase bone mass in men with primary or hypogonadal osteoporosis at high risk for fracture or who have failed or are intolerant to other available osteoporosis therapy.
- For the treatment of men and women with osteoporosis associated with sustained systemic glucocorticoid therapy (daily dosage equivalent to 5 mg or greater of prednisone) at high risk for fracture or who have failed or are intolerant to other available osteoporosis therapy.
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dosage is 20 mcg per dose given subcutaneously once a day. Instruct patients to take supplemental calcium and vitamin D if daily dietary intake is inadequate.
2.2 Administration Instructions
- Administer FORTEO as a subcutaneous injection into the thigh or abdominal region. FORTEO is not approved for intravenous or intramuscular use.
- FORTEO should be administered initially under circumstances in which the patient can sit or lie down if symptoms of orthostatic hypotension occur [see Warnings and Precautions (5.4)].
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration (FORTEO is a clear and colorless liquid). Do not use if solid particles appear or if the solution is cloudy or colored.
- Patients and/or caregivers who administer FORTEO should receive appropriate training and instruction on the proper use of the FORTEO prefilled delivery device (pen) from a qualified health professional.
- Discard the delivery device 28 days after first use.
2.3 Recommended Treatment Duration
Use of FORTEO for more than 2 years during a patient's lifetime should only be considered if a patient remains at or has returned to having a high risk for fracture [see Warnings and Precautions (5.1)].
3 DOSAGE FORMS AND STRENGTHS
Injection: 560 mcg/2.24 mL (250 mcg/mL) clear, colorless solution in a single-patient-use prefilled delivery device (pen) intended to deliver 28 daily doses of 20 mcg.
4 CONTRAINDICATIONS
FORTEO is contraindicated in patients with hypersensitivity to teriparatide or to any of its excipients. Hypersensitivity reactions have included angioedema and anaphylaxis [see Adverse Reactions (6.3)].
5 WARNINGS AND PRECAUTIONS
5.1 Osteosarcoma
An increase in the incidence of osteosarcoma (a malignant bone tumor) was observed in male and female rats treated with teriparatide. Osteosarcoma has been reported in patients treated with FORTEO in the post marketing setting; however, an increased risk of osteosarcoma has not been observed in observational studies in humans. There are limited data assessing the risk of osteosarcoma beyond 2 years of FORTEO use [see Dosage and Administration (2.3), Adverse Reactions (6.3), and Nonclinical Toxicology (13.1)].
Avoid FORTEO use in patients with (these patients are at increased baseline risk of osteosarcoma):
- Open epiphyses (pediatric and young adult patients) (FORTEO is not approved in pediatric patients) [see Use in Specific Populations (8.4)].
- Metabolic bone diseases other than osteoporosis, including Paget's disease of the bone.
- Bone metastases or a history of skeletal malignancies.
- Prior external beam or implant radiation therapy involving the skeleton.
- Hereditary disorders predisposing to osteosarcoma.
5.2 Hypercalcemia and Cutaneous Calcification
Hypercalcemia
FORTEO has not been studied in patients with pre-existing hypercalcemia. FORTEO may cause hypercalcemia and may exacerbate hypercalcemia in patients with pre-existing hypercalcemia [see Adverse Reactions (6.1, 6.3)]. Avoid FORTEO in patients known to have an underlying hypercalcemic disorder, such as primary hyperparathyroidism.
Risk of Cutaneous Calcification Including Calciphylaxis
Serious reports of calciphylaxis and worsening of previously stable cutaneous calcification have been reported in the post-marketing setting in patients taking FORTEO. Risk factors for development of calciphylaxis include underlying auto-immune disease, kidney failure, and concomitant warfarin or systemic corticosteroid use. Discontinue FORTEO in patients who develop calciphylaxis or worsening of previously stable cutaneous calcification.
5.3 Risk of Urolithiasis
In clinical trials, the frequency of urolithiasis was similar in patients treated with FORTEO and patients treated with placebo. However, FORTEO has not been studied in patients with active urolithiasis. If FORTEO-treated patients have pre-existing hypercalciuria or suspected/known active urolithiasis, consider measuring urinary calcium excretion. Consider the risks and benefits of use in patients with active or recent urolithiasis because of the potential to exacerbate this condition.
5.4 Orthostatic Hypotension
FORTEO should be administered initially under circumstances in which the patient can sit or lie down if symptoms of orthostatic hypotension occur. In short-term clinical pharmacology studies of FORTEO in healthy volunteers, transient episodes of symptomatic orthostatic hypotension were observed in 5% of volunteers. Typically, these events began within 4 hours of dosing and resolved (without treatment) within a few minutes to a few hours. When transient orthostatic hypotension occurred, it happened within the first several doses, it was relieved by placing the person in a reclining position, and it did not preclude continued treatment.
5.5 Risk of Digoxin Toxicity
Hypercalcemia may predispose patients to digitalis toxicity because FORTEO transiently increases serum calcium. Consider the potential onset of signs and symptoms of digitalis toxicity when FORTEO is used in patients receiving digoxin [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
Men with Primary or Hypogonadal Osteoporosis and Postmenopausal Women with Osteoporosis
The safety of FORTEO in the treatment of osteoporosis in men and postmenopausal women was assessed in two randomized, double-blind, placebo-controlled trials of 1382 patients (21% men, 79% women) aged 28 to 86 years (mean 67 years) [see Clinical Studies (14.1, 14.2)]. The median durations of the trials were 11 months for men and 19 months for women, with 691 patients exposed to FORTEO and 691 patients to placebo. All patients received 1000 mg of calcium plus at least 400 IU of vitamin D supplementation per day.
The incidence of all-cause mortality was 1% in the FORTEO group and 1% in the placebo group. The incidence of serious adverse events was 16% in the FORTEO group and 19% in the placebo group. Early discontinuation due to adverse events occurred in 7% in the FORTEO group and 6% in the placebo group.
Table 1 lists adverse events from these two trials that occurred in ≥2% of FORTEO-treated and more frequently than placebo-treated patients.
| FORTEO N=691 |
Placebo N=691 |
|
| Event Classification | (%) | (%) |
| Body as a Whole | ||
| Pain | 21.3 | 20.5 |
| Headache | 7.5 | 7.4 |
| Asthenia | 8.7 | 6.8 |
| Neck pain | 3.0 | 2.7 |
| Cardiovascular | ||
| Hypertension | 7.1 | 6.8 |
| Angina pectoris | 2.5 | 1.6 |
| Syncope | 2.6 | 1.4 |
| Digestive System | ||
| Nausea | 8.5 | 6.7 |
| Constipation | 5.4 | 4.5 |
| Diarrhea | 5.1 | 4.6 |
| Dyspepsia | 5.2 | 4.1 |
| Vomiting | 3.0 | 2.3 |
| Gastrointestinal disorder | 2.3 | 2.0 |
| Tooth disorder | 2.0 | 1.3 |
| Musculoskeletal | ||
| Arthralgia | 10.1 | 8.4 |
| Leg cramps | 2.6 | 1.3 |
| Nervous System | ||
| Dizziness | 8.0 | 5.4 |
| Depression | 4.1 | 2.7 |
| Insomnia | 4.3 | 3.6 |
| Vertigo | 3.8 | 2.7 |
| Respiratory System | ||
| Rhinitis | 9.6 | 8.8 |
| Cough increased | 6.4 | 5.5 |
| Pharyngitis | 5.5 | 4.8 |
| Dyspnea | 3.6 | 2.6 |
| Pneumonia | 3.9 | 3.3 |
| Skin and Appendages | ||
| Rash | 4.9 | 4.5 |
| Sweating | 2.2 | 1.7 |
Laboratory Findings
Serum Calcium — FORTEO transiently increased serum calcium, with the maximal effect observed at approximately 4 to 6 hours post-dose. Serum calcium measured at least 16 hours post-dose was not different from pretreatment levels. In clinical trials, the frequency of at least 1 episode of transient hypercalcemia in the 4 to 6 hours after FORTEO administration was 11% of women and 6% of men treated with FORTEO compared to 2% of women and 0% of the men treated with placebo. The percentage of patients treated with FORTEO whose transient hypercalcemia was verified on consecutive measurements was 3% of women and 1% of men.
Urinary Calcium — FORTEO increased urinary calcium excretion, but the frequency of hypercalciuria in clinical trials was similar for patients treated with FORTEO and placebo [see Clinical Pharmacology (12.2)].
Serum Uric Acid — FORTEO increased serum uric acid concentrations. In clinical trials, 3% of FORTEO-treated patients had serum uric acid concentrations above the upper limit of normal compared with 1% of placebo-treated patients. However, the hyperuricemia did not result in an increase in gout, arthralgia, or urolithiasis.
Men and Women with Glucocorticoid-Induced Osteoporosis
The safety of FORTEO in the treatment of men and women with glucocorticoid-induced osteoporosis was assessed in a randomized, double-blind, active-controlled trial of 428 patients (19% men, 81% women) aged 22 to 89 years (mean 57 years) treated with ≥5mg per day prednisone or equivalent for a minimum of 3 months [see Clinical Studies (14.3)]. The duration of the trial was 18 months with 214 patients exposed to FORTEO and 214 patients exposed to an oral daily bisphosphonate (active control). All patients received 1000 mg of calcium plus 800 IU of vitamin D supplementation per day.
There was no increase in mortality in the FORTEO group compared to the active control group. The incidence of serious adverse events was 21% in FORTEO patients and 18% in active control patients, and included pneumonia (3% FORTEO, 1% active control). Early discontinuation because of adverse events occurred in 15% of FORTEO patients and 12% of active control patients, and included dizziness (2% FORTEO, 0% active control).
Adverse events reported at a higher incidence in the FORTEO group and with at least a 2% difference in FORTEO-treated patients compared with active control-treated patients were: nausea (14%, 7%), gastritis (7%, 3%), pneumonia (6%, 3%), dyspnea (6%, 3%), insomnia (5%, 1%), anxiety (4%, 1%), and herpes zoster (3%, 1%), respectively.
6.2 Immunogenicity
As with all peptides, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies or to other teriparatide products may be misleading.
In the clinical trial of postmenopausal women with osteoporosis [see Clinical Studies (14.1)], antibodies that cross reacted with teriparatide were detected in 3% of women (15/541) who received FORTEO. Generally, antibodies were first detected following 12 months of treatment and diminished after withdrawal of therapy. There was no evidence of hypersensitivity reactions among these patients. Antibody formation did not appear to have effects on serum calcium, or on bone mineral density (BMD) response.
6.3 Postmarketing Experience
Adverse Reactions from Postmarketing Spontaneous Reports
The following adverse reactions have been identified during postapproval use of FORTEO. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Cases of bone tumor and osteosarcoma have been reported rarely in the postmarketing period [see Warnings and Precautions (5.2)].
- Hypercalcemia greater than 13 mg/dL has been reported with FORTEO use.
Adverse events reported since market introduction that were temporally related to FORTEO therapy include the following:
- Allergic Reactions: Anaphylactic reactions, drug hypersensitivity, angioedema, urticaria
- Investigations: Hyperuricemia
- Respiratory System: Acute dyspnea, chest pain
- Musculoskeletal: Muscle spasms of the leg or back
- Other: Injection site reactions including injection site pain, swelling and bruising; oro-facial edema
Adverse Reactions from Observational Studies to Assess Incidence of Osteosarcoma
Two osteosarcoma surveillance safety studies (U.S. claims-based database studies) were designed to obtain data on the incidence rate of osteosarcoma among FORTEO-treated patients. In these two studies, three and zero osteosarcoma cases were identified among 379,283 and 153,316 FORTEO users, respectively. The study results suggest a similar risk for osteosarcoma between FORTEO users and their comparators. However, the interpretation of the study results calls for caution owing to the limitations of the data sources which do not allow for complete measurement and control for confounders.
7 DRUG INTERACTIONS
7.1 Digoxin
Sporadic case reports have suggested that hypercalcemia may predispose patients to digitalis toxicity. FORTEO may transiently increase serum calcium. Consider the potential onset of signs and symptoms of digitalis toxicity when FORTEO is used in patients receiving digoxin [see Warnings and Precaution (5.5) and Clinical Pharmacology (12.3)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on FORTEO use in pregnant women to evaluate for drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. Consider discontinuing FORTEO when pregnancy is recognized.
In animal reproduction studies, teriparatide increased skeletal deviations and variations in mouse offspring at subcutaneous doses equivalent to more than 60 times the recommended 20 mcg human daily dose (based on body surface area, mcg/m2), and produced mild growth retardation and reduced motor activity in rat offspring at subcutaneous doses equivalent to more than 120 times the human dose (see Data).
The background risk of major birth defects and miscarriage for the indicated population is unknown. The background risk in the US general population of major birth defects is 2% to 4% and of miscarriage is 15% to 20% of clinically recognized pregnancies.
Data
Animal Data
In animal reproduction studies, pregnant mice received teriparatide during organogenesis at subcutaneous doses equivalent to 8 to 267 times the human dose (based on body surface area, mcg/m2). At subcutaneous doses ≥60 times the human dose, the fetuses showed an increased incidence of skeletal deviations or variations (interrupted rib, extra vertebra or rib). When pregnant rats received teriparatide during organogenesis at subcutaneous doses 16 to 540 times the human dose, the fetuses showed no abnormal findings.
In a perinatal/postnatal study in pregnant rats dosed subcutaneously from organogenesis through lactation, mild growth retardation was observed in female offspring at doses ≥120 times the human dose. Mild growth retardation in male offspring and reduced motor activity in both male and female offspring were observed at maternal doses of 540 times the human dose. There were no developmental or reproductive effects in mice or rats at doses 8 or 16 times the human dose, respectively.
8.4 Pediatric Use
The safety and effectiveness of FORTEO have not been established in pediatric patients. Pediatric patients are at higher baseline risk of osteosarcoma because of open epiphyses [see Warnings and Precautions (5.1)].
8.5 Geriatric Use
Of the patients who received FORTEO in the osteoporosis trial of 1637 postmenopausal women, 75% were 65 years of age and older and 23% were 75 years of age and older. Of the patients who received FORTEO in the trial of 437 men with primary or hypogonadal osteoporosis, 39% were 65 years of age and over and 13% were 75 years of age and over. Of the 214 patients who received FORTEO in the glucocorticoid induced osteoporosis trial, 28% were 65 years of age and older and 9% were 75 years of age and older. No overall differences in safety or effectiveness of FORTEO have been observed between patients 65 years of age and older and younger adult patients.
8.6 Hepatic Impairment
No studies have been performed in patients with hepatic impairment [see Clinical Pharmacology (12.3)].
8.7 Renal Impairment
In 5 patients with severe renal impairment (CrCl<30 mL/minute), the AUC and T1/2 of teriparatide were increased by 73% and 77%, respectively. Maximum serum concentration of teriparatide was not increased. It is unknown whether FORTEO alters the underlying metabolic bone disease seen in chronic renal impairment [see Clinical Pharmacology (12.3)].
10 OVERDOSAGE
In postmarketing spontaneous reports, there have been cases of medication errors in which the entire contents (up to 800 mcg) (40 times the recommended dose) of the FORTEO prefilled delivery device (pen) have been administered as a single dose. Transient events reported have included nausea, weakness/lethargy and hypotension. No fatalities associated with overdose have been reported. Additional signs, symptoms, and complications of FORTEO overdosage may include a delayed hypercalcemic effect, vomiting, dizziness, and headache.
11 DESCRIPTION
FORTEO (teriparatide injection) is a recombinant human parathyroid hormone analog (PTH 1-34). It has an identical sequence to the 34 N-terminal amino acids (the biologically active region) of the 84-amino acid human parathyroid hormone.
The molecular formula of teriparatide is C181H291N55O51S2 and molecular weight is 4117.8 daltons. Its amino acid sequence is shown below:
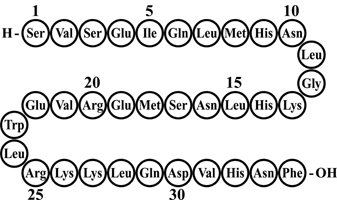
Teriparatide is manufactured using a strain of Escherichia coli modified by recombinant DNA technology.
FORTEO is supplied as a sterile, colorless, clear, isotonic solution in a glass cartridge which is pre-assembled into a single-patient-use delivery device (pen) for subcutaneous injection. Each delivery device (pen) is filled with volume to allow delivery of 2.24 mL. Each mL contains 250 mcg of teriparatide (as a free base), 0.41 mg of glacial acetic acid, 0.1 mg of sodium acetate (anhydrous), 45.4 mg of mannitol, 3 mg of Metacresol, and Water for Injection. In addition, hydrochloric acid solution 10% and/or sodium hydroxide solution 10% may have been added to adjust the pH to 4.
Each prefilled delivery device (pen) delivers 20 mcg of teriparatide per dose for up to 28 days. Each device contains additional volume to allow troubleshooting of the device 2 times.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Endogenous 84-amino acid parathyroid hormone (PTH) is the primary regulator of calcium and phosphate metabolism in bone and kidney. Physiological actions of PTH include regulation of bone metabolism, renal tubular reabsorption of calcium and phosphate, and intestinal calcium absorption. The biological actions of PTH and teriparatide are mediated through binding to specific high-affinity cell-surface receptors. Teriparatide and the 34 N-terminal amino acids of PTH bind to these receptors with the same affinity and have the same physiological actions on bone and kidney. Teriparatide is not expected to accumulate in bone or other tissues.
The skeletal effects of teriparatide depend upon the pattern of systemic exposure. Once-daily administration of teriparatide stimulates new bone formation on trabecular and cortical (periosteal and/or endosteal) bone surfaces by preferential stimulation of osteoblastic activity over osteoclastic activity. In monkey studies, teriparatide improved trabecular microarchitecture and increased bone mass and strength by stimulating new bone formation in both cancellous and cortical bone. In humans, the anabolic effects of teriparatide manifest as an increase in skeletal mass, an increase in markers of bone formation and resorption, and an increase in bone strength. By contrast, continuous excess of endogenous PTH, as occurs in hyperparathyroidism, may be detrimental to the skeleton because bone resorption may be stimulated more than bone formation.
12.2 Pharmacodynamics
Pharmacodynamics in Men with Primary or Hypogonadal Osteoporosis and Postmenopausal Women with Osteoporosis
Effects on Mineral Metabolism — Teriparatide affects calcium and phosphorus metabolism in a pattern consistent with the known actions of endogenous PTH (e.g., increases serum calcium and decreases serum phosphorus).
Serum Calcium Concentrations — When teriparatide 20 mcg was administered once daily, the serum calcium concentration increased transiently, beginning approximately 2 hours after dosing and reaching a maximum concentration between 4 and 6 hours (median increase, 0.4 mg/dL). The serum calcium concentration began to decline approximately 6 hours after dosing and returned to baseline by 16 to 24 hours after each dose.
In a clinical study of postmenopausal women with osteoporosis, the median peak serum calcium concentration measured 4 to 6 hours after dosing with FORTEO (20 mcg subcutaneous once daily) was 9.68 mg/dL at 12 months. The peak serum calcium remained below 11 mg/dL in >99% of women at each visit. Sustained hypercalcemia was not observed.
In this study, 11.1% of women treated with FORTEO had at least 1 serum calcium value above the upper limit of normal (ULN) (10.6 mg/dL) compared with 1.5% of women treated with placebo. The percentage of women treated with FORTEO whose serum calcium was above the ULN on consecutive 4- to 6-hour post-dose measurements was 3% compared with 0.2% of women treated with placebo. In these women, calcium supplements and/or FORTEO doses were reduced. The timing of these dose reductions was at the discretion of the investigator. FORTEO dose adjustments were made at varying intervals after the first observation of increased serum calcium (median 21 weeks). During these intervals, there was no evidence of progressive increases in serum calcium.
In a clinical study of men with either primary or hypogonadal osteoporosis, the effects on serum calcium were similar to those observed in postmenopausal women. The median peak serum calcium concentration measured 4 to 6 hours after dosing with FORTEO was 9.44 mg/dL at 12 months. The peak serum calcium remained below 11 mg/dL in 98% of men at each visit. Sustained hypercalcemia was not observed.
In this study, 6% of men treated with FORTEO daily had at least 1 serum calcium value above the ULN (10.6 mg/dL) compared with none of the men treated with placebo. The percentage of men treated with FORTEO whose serum calcium was above the ULN on consecutive measurements was 1.3% (2 men) compared with none of the men treated with placebo. Calcium supplementation was reduced in these men [see Warnings and Precautions (5.2) and Adverse Reactions (6.1)].
In a clinical study of women previously treated for 18 to 39 months with raloxifene (n=26) or alendronate (n=33), mean serum calcium >12 hours after FORTEO treatment was increased by 0.36 to 0.56 mg/dL, after 1 to 6 months of FORTEO treatment compared with baseline. Of the women pretreated with raloxifene, 3 (11.5%) had a serum calcium >11 mg/dL, and of those pretreated with alendronate, 3 (9.1%) had a serum calcium >11 mg/dL. The highest serum calcium reported was 12.5 mg/dL. None of the women had symptoms of hypercalcemia. There were no placebo controls in this study.
In the study of patients with glucocorticoid-induced osteoporosis, the effects of FORTEO on serum calcium were similar to those observed in postmenopausal women with osteoporosis not taking glucocorticoids.
Urinary Calcium Excretion — In a clinical study of postmenopausal women with osteoporosis who received 1000 mg of supplemental calcium and at least 400 IU of vitamin D, daily FORTEO increased urinary calcium excretion. The median urinary excretion of calcium was 190 mg/day at 6 months and 170 mg/day at 12 months. These levels were 30 mg/day and 12 mg/day higher, respectively, than in women treated with placebo. The incidence of hypercalciuria (>300 mg/day) was similar in the women treated with FORTEO or placebo.
In a clinical study of men with either primary or hypogonadal osteoporosis who received 1000 mg of supplemental calcium and at least 400 IU of vitamin D, daily FORTEO had inconsistent effects on urinary calcium excretion. The median urinary excretion of calcium was 220 mg/day at 1 month and 210 mg/day at 6 months. These levels were 20 mg/day higher and 8 mg/day lower, respectively, than in men treated with placebo. The incidence of hypercalciuria (>300 mg/day) was similar in the men treated with FORTEO or placebo.
Phosphorus and Vitamin D — In single-dose studies, teriparatide produced transient phosphaturia and mild transient reductions in serum phosphorus concentration. However, hypophosphatemia (<2.4 mg/dL) was not observed in clinical trials with FORTEO.
In clinical trials of daily FORTEO, the median serum concentration of 1,25-dihydroxyvitamin D was increased at 12 months by 19% in women and 14% in men, compared with baseline. In the placebo group, this concentration decreased by 2% in women and increased by 5% in men. The median serum 25-hydroxyvitamin D concentration at 12 months was decreased by 19% in women and 10% in men compared with baseline. In the placebo group, this concentration was unchanged in women and increased by 1% in men.
In the study of patients with glucocorticoid-induced osteoporosis, the effects of FORTEO on serum phosphorus were similar to those observed in postmenopausal women with osteoporosis not taking glucocorticoids.
Effects on Markers of Bone Turnover — Daily administration of FORTEO to men and postmenopausal women with osteoporosis in clinical studies stimulated bone formation, as shown by increases in the formation markers serum bone-specific alkaline phosphatase (BSAP) and procollagen I carboxy-terminal propeptide (PICP). Data on biochemical markers of bone turnover were available for the first 12 months of treatment. Peak concentrations of PICP at 1 month of treatment were approximately 41% above baseline, followed by a decline to near-baseline values by 12 months. BSAP concentrations increased by 1 month of treatment and continued to rise more slowly from 6 through 12 months. The maximum increases of BSAP were 45% above baseline in women and 23% in men. After discontinuation of therapy, BSAP concentrations returned toward baseline. The increases in formation markers were accompanied by secondary increases in the markers of bone resorption: urinary N-telopeptide (NTX) and urinary deoxypyridinoline (DPD), consistent with the physiological coupling of bone formation and resorption in skeletal remodeling. Changes in BSAP, NTX, and DPD were lower in men than in women, possibly because of lower systemic exposure to teriparatide in men.
In the study of patients with glucocorticoid-induced osteoporosis, the effects of FORTEO on serum markers of bone turnover were similar to those observed in postmenopausal women with osteoporosis not taking glucocorticoids.
12.3 Pharmacokinetics
Absorption — Teriparatide is absorbed after subcutaneous injection; the absolute bioavailability is approximately 95% based on pooled data from 20-, 40-, and 80- mcg doses (1-, 2-, and 4- times the recommended dosage, respectively). The peptide reaches peak serum concentrations about 30 minutes after subcutaneous injection of a 20-mcg dose and declines to non-quantifiable concentrations within 3 hours.
Elimination — Systemic clearance of teriparatide (approximately 62 L/hour in women and 94 L/hour in men) exceeds the rate of normal liver plasma flow, consistent with both hepatic and extra-hepatic clearance. The half-life of teriparatide in serum was approximately 1 hour when administered by subcutaneous injection.
No metabolism or excretion studies have been performed with teriparatide. Peripheral metabolism of PTH is believed to occur by non-specific enzymatic mechanisms in the liver followed by excretion via the kidneys.
Specific Populations
Geriatric Patients — No age-related differences in teriparatide pharmacokinetics were detected (range 31 to 85 years).
Male and Female Patients — Although systemic exposure to teriparatide was approximately 20% to 30% lower in men than women, the recommended dosage for men and women is the same.
Patients with Renal Impairment — No pharmacokinetic differences were identified in 11 patients with creatinine clearance (CrCl) 30 to 72 mL/minute administered a single dose of teriparatide. In 5 patients with severe renal impairment (CrCl<30 mL/minute), the AUC and T1/2 of teriparatide were increased by 73% and 77%, respectively. Maximum serum concentration of teriparatide was not increased. No studies have been performed in patients undergoing dialysis for chronic renal failure.
Drug Interaction Studies
Digoxin — In a study of 15 healthy people administered digoxin daily to steady state, a single FORTEO dose did not alter the effect of digoxin on the systolic time interval (from electrocardiographic Q-wave onset to aortic valve closure, a measure of digoxin's calcium-mediated cardiac effect).
Hydrochlorothiazide — In a study of 20 healthy people, the coadministration of hydrochlorothiazide 25 mg with 40 mcg of FORTEO (2 times the recommended dose) did not affect the serum calcium response to FORTEO. The 24-hour urine excretion of calcium was reduced by a clinically unimportant amount (15%). The effect of coadministration of a higher dose of hydrochlorothiazide with FORTEO on serum calcium levels has not been studied.
Furosemide — In a study of 9 healthy people and 17 patients with CrCl 13 to 72 mL/minute, coadministration of intravenous furosemide (20 to 100 mg) with FORTEO 40 mcg (2 times the recommended dose) resulted in small increases in the serum calcium (2%) and 24-hour urine calcium (37%); however, these changes did not appear to be clinically important.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Two carcinogenicity bioassays were conducted in Fischer 344 rats. In the first study, male and female rats were given daily subcutaneous teriparatide injections of 5, 30, or 75 mcg/kg/day for 24 months from 2 months of age. These doses resulted in rat systemic exposures that were 3, 20, and 60 times higher than the systemic exposure observed in humans, respectively, following a subcutaneous dose of 20 mcg (based on AUC comparison). Teriparatide treatment resulted in a marked dose-related increase in the incidence of osteosarcoma, a rare malignant bone tumor, in both male and female rats. Osteosarcomas were observed at all doses and the incidence reached 40% to 50% in the high-dose groups. Teriparatide also caused a dose-related increase in osteoblastoma and osteoma in both sexes. No osteosarcomas, osteoblastomas or osteomas were observed in untreated control rats. The bone tumors in rats occurred in association with a large increase in bone mass and focal osteoblast hyperplasia.
The second 2-year study was carried out in order to determine the effect of treatment duration and animal age on the development of bone tumors. Female rats were treated for different periods between 2 and 26 months of age with subcutaneous teriparatide doses of 5 and 30 mcg/kg (equivalent to 3 and 20 times the human exposure at the 20-mcg dose, respectively, based on AUC comparison). The study showed that the occurrence of osteosarcoma, osteoblastoma and osteoma was dependent upon dose and duration of teriparatide exposure. Bone tumors were observed when immature 2-month old rats were treated with 30 mcg/kg/day of teriparatide for 24 months or with 5 or 30 mcg/kg/day of teriparatide for 6 months. Bone tumors were also observed when mature 6-month old rats were treated with 30 mcg/kg/day of teriparatide for 6 or 20 months. Tumors were not detected when mature 6-month old rats were treated with 5 mcg/kg/day of teriparatide for 6 or 20 months. The results did not demonstrate a difference in susceptibility to bone tumor formation, associated with teriparatide treatment, between mature and immature rats.
No bone tumors were detected in a long-term monkey study [see Nonclinical Toxicology (13.2)].
Mutagenesis
Teriparatide was not genotoxic in any of the following test systems: the Ames test for bacterial mutagenesis; the mouse lymphoma assay for mammalian cell mutation; the chromosomal aberration assay in Chinese hamster ovary cells, with and without metabolic activation; and the in vivo micronucleus test in mice.
13.2 Animal Toxicology
In single-dose rodent studies using subcutaneous injection of teriparatide, no mortality was seen in rats given doses of 1000 mcg/kg (540 times the human dose based on surface area, mcg/m2) or in mice given 10,000 mcg/kg (2700 times the human dose based on surface area, mcg/m2).
In a long-term study, skeletally mature ovariectomized female monkeys (N=30 per treatment group) were given either daily subcutaneous teriparatide injections of 5 mcg/kg or vehicle. Following the 18-month treatment period, the monkeys were removed from teriparatide treatment and were observed for an additional 3 years. The 5 mcg/kg dose resulted in systemic exposures that were approximately 6 times higher than the systemic exposure observed in humans following a subcutaneous dose of 20 mcg (based on AUC comparison). Bone tumors were not detected by radiographic or histologic evaluation in any monkey in the study.
14 CLINICAL STUDIES
14.1 Treatment of Osteoporosis in Postmenopausal Women
The safety and efficacy of once-daily FORTEO, median exposure of 19 months, were examined in a double-blind, multicenter, placebo-controlled clinical study of 1637 postmenopausal women with osteoporosis. In this study 541 postmenopausal women were treated with 20 mcg FORTEO subcutaneously once daily.
All women received 1000 mg of calcium and at least 400 IU of vitamin D per day. Baseline and endpoint spinal radiographs were evaluated using the semiquantitative scoring. Ninety percent of the women in the study had 1 or more radiographically diagnosed vertebral fractures at baseline. The primary efficacy endpoint was the occurrence of new radiographically diagnosed vertebral fractures defined as changes in the height of previously undeformed vertebrae. Such fractures are not necessarily symptomatic.
Effect on Fracture Incidence
New Vertebral Fractures — FORTEO, when taken with calcium and vitamin D and compared with calcium and vitamin D alone, reduced the risk of 1 or more new vertebral fractures from 14.3% of women in the placebo group to 5.0% in the FORTEO group (444 of the 541 patients treated with 20 mcg once daily of FORTEO were included in this analysis). This difference was statistically significant (p<0.001); the absolute reduction in risk was 9.3% and the relative reduction was 65%. FORTEO was effective in reducing the risk for vertebral fractures regardless of age, baseline rate of bone turnover, or baseline BMD (see Table 2).
|
a p≤0.001 compared with placebo. |
||||
| Percent of Women With Fracture | ||||
FORTEO (N=444) |
Placebo (N=448) |
Absolute Risk Reduction (%, 95% CI) |
Relative Risk Reduction (%, 95% CI) |
|
| New fracture (≥1) | 5.0a | 14.3 | 9.3 (5.5-13.1) | 65 (45-78) |
| 1 fracture | 3.8 | 9.4 | ||
| 2 fractures | 0.9 | 2.9 | ||
| ≥3 fractures | 0.2 | 2.0 | ||
New Nonvertebral Osteoporotic Fractures — FORTEO significantly reduced the risk of any nonvertebral fracture from 5.5% in the placebo group to 2.6% in the FORTEO group (p<0.05). The absolute reduction in risk was 2.9% and the relative reduction was 53%. The incidence of new nonvertebral fractures in the FORTEO group compared with the placebo group was ankle/foot (0.2%, 0.7%), hip (0.2%, 0.7%), humerus (0.4%, 0.4%), pelvis (0%, 0.6%), ribs (0.6%, 0.9%), wrist (0.4%, 1.3%), and other sites (1.1%, 1.5%), respectively.
The cumulative percentage of postmenopausal women with osteoporosis who sustained new nonvertebral fractures was lower in women treated with FORTEO than in women treated with placebo (see Figure 1).
Figure 1: Cumulative Percentage of Postmenopausal Women with Osteoporosis Sustaining New Nonvertebral Osteoporotic Fractures
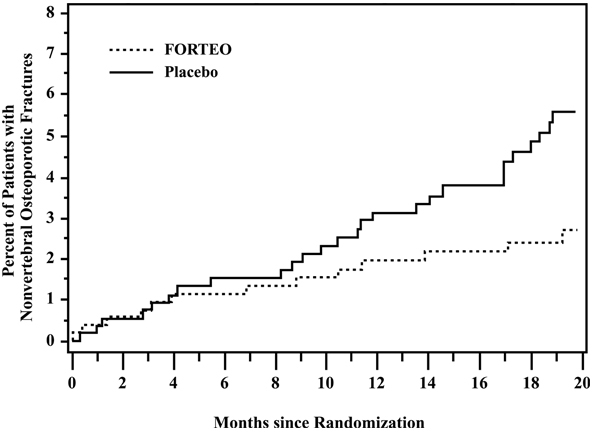
Effect on Bone Mineral Density (BMD)
FORTEO increased lumbar spine BMD in postmenopausal women with osteoporosis. Statistically significant increases were seen at 3 months and continued throughout the treatment period. Postmenopausal women with osteoporosis who were treated with FORTEO had statistically significant increases in BMD from baseline to endpoint at the lumbar spine, femoral neck, total hip, and total body (see Table 3).
|
a Intent-to-treat analysis, last observation carried forward. |
||
|
b p<0.001 compared with placebo. |
||
|
c p<0.05 compared with placebo. |
||
| FORTEO N=541 |
Placebo N=544 |
|
| Lumbar spine BMD | 9.7b | 1.1 |
| Femoral neck BMD | 2.8c | -0.7 |
| Total hip BMD | 2.6c | -1.0 |
| Trochanter BMD | 3.5c | -0.2 |
| Intertrochanter BMD | 2.6c | -1.3 |
| Ward's triangle BMD | 4.2c | -0.8 |
| Total body BMD | 0.6c | -0.5 |
| Distal 1/3 radius BMD | -2.1 | -1.3 |
| Ultradistal radius BMD | -0.1 | -1.6 |
FORTEO treatment increased lumbar spine BMD from baseline in 96% of postmenopausal women treated. Seventy-two percent of patients treated with FORTEO achieved at least a 5% increase in spine BMD, and 44% gained 10% or more.
Both treatment groups lost height during the trial. The mean decreases were 3.61 and 2.81 mm in the placebo and FORTEO groups, respectively.
Bone Histology
The effects of FORTEO on bone histology were evaluated in iliac crest biopsies of 35 postmenopausal women treated for 12 to 24 months with calcium and vitamin D and FORTEO. Normal mineralization was observed with no evidence of cellular toxicity. The new bone formed with FORTEO was of normal quality (as evidenced by the absence of woven bone and marrow fibrosis).
14.2 Treatment to Increase Bone Mass in Men with Primary or Hypogonadal Osteoporosis
The safety and efficacy of once-daily FORTEO, median exposure of 10 months, were examined in a double-blind, multicenter, placebo-controlled clinical study of 437 men with either primary (idiopathic) or hypogonadal osteoporosis. In this study, 151 men received 20 mcg of FORTEO given subcutaneously once daily. All men received 1000 mg of calcium and at least 400 IU of vitamin D per day. The primary efficacy endpoint was change in lumbar spine BMD.
FORTEO increased lumbar spine BMD in men with primary or hypogonadal osteoporosis. Statistically significant increases were seen at 3 months and continued throughout the treatment period. FORTEO was effective in increasing lumbar spine BMD regardless of age, baseline rate of bone turnover, and baseline BMD. The effects of FORTEO at additional skeletal sites are shown in Table 4.
FORTEO treatment for a median of 10 months increased lumbar spine BMD from baseline in 94% of men treated. Fifty-three percent of patients treated with FORTEO achieved at least a 5% increase in spine BMD, and 14% gained 10% or more.
|
a Intent-to-treat analysis, last observation carried forward. |
||
|
b p<0.001 compared with placebo. |
||
|
c p<0.05 compared with placebo. |
||
| FORTEO N=151 |
Placebo N=147 |
|
| Lumbar spine BMD | 5.9b | 0.5 |
| Femoral neck BMD | 1.5c | 0.3 |
| Total hip BMD | 1.2 | 0.5 |
| Trochanter BMD | 1.3 | 1.1 |
| Intertrochanter BMD | 1.2 | 0.6 |
| Ward's triangle BMD | 2.8 | 1.1 |
| Total body BMD | 0.4 | -0.4 |
| Distal 1/3 radius BMD | -0.5 | -0.2 |
| Ultradistal radius BMD | -0.5 | -0.3 |
14.3 Treatment of Men and Women with Glucocorticoid-Induced Osteoporosis
The efficacy of FORTEO for treating glucocorticoid-induced osteoporosis was assessed in a randomized, double-blind, active-controlled trial of 428 patients (19% men, 81% women) aged 22 to 89 years (mean 57 years) treated with ≥5 mg/day prednisone or equivalent for a minimum of 3 months. The duration of the trial was 18 months. In the trial 214 patients were treated with FORTEO 20 mcg given subcutaneously once daily. In the FORTEO group, the baseline median glucocorticoid dose was 7.5 mg/day and the baseline median duration of glucocorticoid use was 1.5 years. The mean (SD) baseline lumbar spine BMD was 0.85 ± 0.13 g/cm2 and lumbar spine BMD T-score was –2.5 ± 1 (number of standard deviations below the mean BMD value for healthy adults). A total of 30% of patients had prevalent vertebral fracture(s) and 43% had prior non-vertebral fracture(s). The patients had chronic rheumatologic, respiratory or other diseases that required sustained glucocorticoid therapy. All patients received 1000 mg of calcium plus 800 IU of vitamin D supplementation per day.
Because of differences in mechanism of action (anabolic vs. anti-resorptive) and lack of clarity regarding differences in BMD as an adequate predictor of fracture efficacy, data on the active comparator are not presented.
Effect on Bone Mineral Density (BMD)
In patients with glucocorticoid-induced osteoporosis, FORTEO increased lumbar spine BMD compared with baseline at 3 months through 18 months of treatment. In patients treated with FORTEO, the mean percent change in BMD from baseline to endpoint was 7.2% at the lumbar spine, 3.6% at the total hip, and 3.7% at the femoral neck (p <0.001 all sites). The relative treatment effects of FORTEO were consistent in subgroups defined by gender, age, geographic region, body mass index, underlying disease, prevalent vertebral fracture, baseline glucocorticoid dose, prior bisphosphonate use, and glucocorticoid discontinuation during trial.
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
FORTEO (teriparatide injection) is a clear and colorless solution, available as single-patient-use prefilled delivery device (pen) in the following package size:
- 560 mcg/2.24 mL (250 mcg/mL) [intended to deliver 28 daily doses of 20 mcg] NDC 0002-9678-01 (MS8400).
16.2 Storage and Handling
- Store FORTEO under refrigeration at 2° to 8°C (36° to 46°F) at all times except when administering the product.
- Recap the delivery device (pen) when not in use to protect the cartridge from physical damage and light.
- When using FORTEO, minimize the time out of the refrigerator; deliver the dose immediately following removal from the refrigerator.
- Do not freeze. Do not use FORTEO if it has been frozen.
- Throw away the device 28 days after first use.
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide and the User Manual) before starting FORTEO and each time the prescription is renewed. Failure to follow the instructions may result in inaccurate dosing.
Osteosarcoma
Patients should be made aware that in rats, teriparatide caused an increase in the incidence of osteosarcoma (a malignant bone tumor). Although cases of osteosarcoma have been reported in patients using FORTEO no increased risk of osteosarcoma was observed in adult humans treated with FORTEO [see Warnings and Precautions (5.1)].
Hypercalcemia
Instruct patients taking FORTEO to contact a health care provider if they develop persistent symptoms of hypercalcemia (e.g., nausea, vomiting, constipation, lethargy, muscle weakness) [see Warnings and Precautions (5.2)].
Orthostatic Hypotension
When initiating FORTEO treatment, instruct patients to be prepared to immediately sit or lie down during or after administration in case they feel lightheaded or have palpitations after the injection. Instruct patients to sit or lie down until the symptoms resolve. If symptoms persist or worsen, instruct patients to consult a healthcare provider before continuing treatment [see Warnings and Precautions (5.4)].
Other Osteoporosis Treatment Modalities
Patients should be informed regarding the roles of supplemental calcium and/or vitamin D.
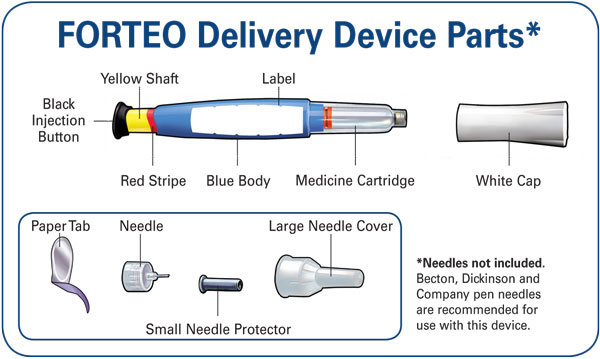
Use of the Prefilled Delivery Device (Pen)
Instruct patients and caregivers who administer FORTEO on how to properly use the delivery device (refer to User Manual), to properly dispose of needles, and not to share their prefilled delivery device with other patients. Instruct patients and caregivers who administer FORTEO that the contents of the delivery device should not be transferred to a syringe.
Inform patients that each FORTEO delivery device can be used up to 28 days after first use. After the 28-day use period, instruct patients to discard the FORTEO delivery device, even if it still contains some unused solution. Instruct patients not to use FORTEO after the expiration date printed on the delivery device and packaging.
Marketed by: Lilly USA, LLC
Indianapolis, IN 46285, USA
www.forteo.com
Pat.: www.lilly.com/patents
Copyright © 2002, 2025, Eli Lilly and Company. All rights reserved.
FOR-0006-USPI-20250603
|
This Medication Guide has been approved by the U.S. Food and Drug Administration. |
Revised: 11/2020 |
| MEDICATION GUIDE FORTEO® (for-TAY-o) teriparatide injection for subcutaneous use |
|
| Read this Medication Guide before you start using FORTEO and each time you get a refill. There may be new information. Also, read the User Manual that comes with the FORTEO delivery device (pen) for information on how to use the device to inject your medicine the right way. This Medication Guide does not take the place of talking with your healthcare provider about your medical condition or your treatment. | |
|
What is the most important information I should know about FORTEO? |
|
| What is FORTEO? FORTEO is a prescription medicine used to:
|
|
| It is not known if FORTEO is safe and effective in children. FORTEO should not be used in children and young adults whose bones are still growing. |
|
| Who should not use FORTEO? Do not use FORTEO if you:
|
|
| Symptoms of a serious allergic reaction of FORTEO may include swelling of the face, lips, tongue or throat that may cause difficulty in breathing or swallowing. Call your healthcare provider right away or get emergency medical help if you get any of these symptoms. | |
| What should I tell my healthcare provider before using FORTEO? Before you use FORTEO, tell your healthcare provider about all of your medical conditions, including if you:
|
|
| Tell your healthcare provider about all the medicines you take including prescription and over-the-counter medicines, vitamins, and herbal supplements. Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine. |
|
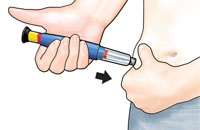
How should I use FORTEO?
|
|
| If your healthcare provider recommends calcium and vitamin D supplements, you can take them at the same time you take FORTEO. | |
| What are the possible side effects of FORTEO? FORTEO may cause serious side effects including:
|
|
The most common side effects of FORTEO include:
|
|
| These are not all the possible side effects of FORTEO. For more information, ask your
healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
How should I store FORTEO?
|
|
| Keep FORTEO and all medicines out of the reach of children. | |
| General information about the safe and effective use of FORTEO. Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use FORTEO for a condition for which it was not prescribed. Do not give FORTEO to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about FORTEO that is written for health professionals. |
|
| What are the ingredients in FORTEO? Active ingredient: teriparatide Inactive ingredients: glacial acetic acid, sodium acetate (anhydrous), mannitol, metacresol, and water for injection. In addition, hydrochloric acid solution 10% and/or sodium hydroxide solution 10% may have been added to adjust the product to pH 4. |
|
| For more information, go to www.FORTEO.com or call Lilly at 1-866-436-7836. Marketed by: Lilly USA, LLC, Indianapolis, IN 46285, USA Copyright © 2002, 2020, Eli Lilly and Company. All rights reserved. FOR-0003-MG-20201116 |
|
FORTEO® (for-TAY-o)
teriparatide injection
User Manual
Important: First read the Medication Guide that comes inside your FORTEO carton.
Before you use your new FORTEO delivery device, please read the entire front and back of this User Manual completely. Follow the directions carefully when using the FORTEO delivery device.
Do not share your delivery device or needles because infection or disease can be spread from one person to another.
The FORTEO delivery device contains 28 days of medicine. Throw away the FORTEO delivery device 28 days after first use, even if it is not completely empty. Do not inject more than one dose of FORTEO in the same day.
Do not transfer FORTEO to a syringe.
Wash your hands before every injection. Prepare the injection site as your healthcare provider instructed.
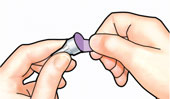
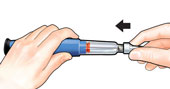
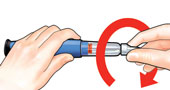
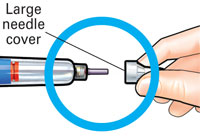
| 2 Attach new needle |
 |
 |
 |
 |
| Pull off paper tab. |
Push needle straight onto medicine cartridge. |
Screw on needle clockwise until firmly attached. |
Pull off large needle cover and save it. |
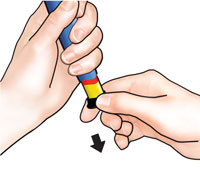
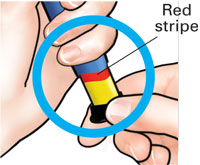
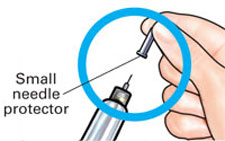
| 3 Set dose |
 |
 |
 |
| Pull out black injection button until it stops. If you cannot pull out the black injection button see Troubleshooting, Problem E, on back page. |
Check to make sure red stripe shows. |
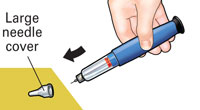
Pull off small needle protector and throw away. |
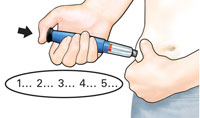
| IMPORTANT | ||||
| 5 Confirm dose |
 |
After completing the injection: Once the needle is removed from the skin, take your thumb off the black injection button. Check to make sure the black injection button is all the way in. If the yellow shaft does not show, you have finished the injection steps the right way. |
 |
You should NOT see any of the yellow shaft. If you do and have already injected the medicine, do not inject yourself a second time on the same day. Instead, you MUST reset the FORTEO delivery device (see Troubleshooting, Problem A, on back page). |

For more information, or if you have any questions, turn to the back of this page.
|
FORTEO® (for-TAY-o) |
|||
| Troubleshooting | |||
| Problem | Solution | ||
|
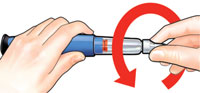
A.
The yellow shaft is still showing |
To reset the FORTEO delivery device, follow the steps below.
|
||
| You can prevent this problem by always using a NEW needle for each injection, and by pushing the black injection button
all the way in and slowly counting to five. |
|||
| B. How can I tell if my FORTEO delivery device works? | The black injection button should be all the way in to show that the full dose of
medicine has been injected from the FORTEO delivery device.
Use a new needle every time you inject to be sure your FORTEO delivery device will work properly. |
||
| C. I see an air bubble in my FORTEO delivery device. | A small air bubble will not affect your dose and it will not harm you. You can continue to take your dose as usual. | ||
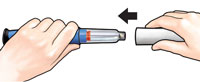
D. I cannot get the needle off. |
|
||
|
E. What should I do if I have difficulty pulling out the black injection button? |
Change to a new FORTEO delivery device to take your dose as instructed by your healthcare
provider. When the black injection button becomes hard to pull out, this means there is not enough medicine in your FORTEO delivery device for another dose. You may still see some medicine left in the cartridge. |
||


| Cleaning and Storage |
Cleaning Your FORTEO Delivery Device
Storing Your FORTEO Delivery Device
|
| Other Important Notes |
|
| Disposal of Pen Needles and Delivery Device | |
Disposal of Pen Needles and the FORTEO Delivery Device
|
|
| Dispose of the FORTEO delivery device 28 days after first use. | 1st use date ______ / ______ / ______ Throw away after ______ / ______ / ______ |
| If you have questions or need help with your FORTEO delivery device, contact Eli Lilly
and Company at 1-800-LillyRx (1-800-545-5979) or your healthcare provider. For more information about FORTEO, go to www.FORTEO.com. Marketed by: Lilly USA, LLC Indianapolis, IN 46285, USA FORTEO is a registered trademark of Eli Lilly and Company. ® Registered trademarks owned by Eli Lilly and Company; used under license. Copyright © 2008, 2025, Eli Lilly and Company. All rights reserved. |
Literature revised Jun 03, 2025
FOR-0004-IFU-20250603
PACKAGE LABEL – FORTEO 20 mcg per dose, 2.24 mL
Do NOT transfer contents to a syringe
ATTENTION PHARMACIST: Medication Guide and device User Manual for patient inside carton
NDC 0002-8400-01
MS8400
FORTEO®
teriparatide injection
20 mcg per dose (given once daily subcutaneously)
Each single-patient-use prefilled pen will deliver 28 subcutaneous doses.
560 mcg/ 2.24 mL (250 mcg/mL)
For Single-Patient-Use Only
REFRIGERATE / DO NOT FREEZE
For subcutaneous use / Rx only
Needles not included
Becton, Dickinson and Company pen needles are recommended for use with this device
www.forteo.com
Lilly

PACKAGE LABEL – FORTEO 20 mcg per dose, 2.24 mL
NDC 0002-9678-01
Do NOT transfer contents to a syringe
ATTENTION PHARMACIST: Medication Guide and device User Manual for patient inside carton
MS8400
FORTEO®
teriparatide injection
20 mcg per dose (given once daily subcutaneously)
Each single-patient-use prefilled pen will deliver 28 subcutaneous doses.
560 mcg/ 2.24 mL (250 mcg/mL)
For Single-Patient-Use Only
REFRIGERATE / DO NOT FREEZE
For subcutaneous use / Rx only
Needles not included
Becton, Dickinson and Company pen needles are recommended for use with this device
www.forteo.com
Lilly
