FULL PRESCRIBING INFORMATION
WARNING: SUICIDAL THOUGHTS AND BEHAVIORS
Antidepressants increased the risk of suicidal thoughts and behavior in children, adolescents, and young adults in short-term studies. These studies did not show an increase in the risk of suicidal thoughts and behavior with antidepressant use in patients over age 24; there was a reduction in risk with antidepressant use in patients aged 65 and older [see Warnings and Precautions (5.1)].
In patients of all ages who are started on antidepressant therapy, monitor closely for worsening, and for emergence of suicidal thoughts and behaviors. Advise families and caregivers of the need for close observation and communication with the prescriber [see Warnings and Precautions (5.1)].
1 INDICATIONS AND USAGE
CYMBALTA® is indicated for the treatment of:
- Major depressive disorder in adults
- Generalized anxiety disorder in adults and pediatric patients 7 years of age and older
- Diabetic peripheral neuropathic pain in adults
- Fibromyalgia in adults and pediatric patients 13 years of age and older
- Chronic musculoskeletal pain in adults
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Instructions
Administer CYMBALTA orally (with or without meals) and swallow whole. Do not chew or crush, and do not open the delayed-release capsule and sprinkle its contents on food or mix with liquids because these actions might affect the enteric coating. If a dose of CYMBALTA is missed, take the missed dose as soon as it is remembered. If it is almost time for the next dose, skip the missed dose and take the next dose at the regular time. Do not take two doses of CYMBALTA at the same time.
2.2 Dosage for Treatment of Major Depressive Disorder in Adults
The recommended starting dosage in adults with MDD is 40 mg/day (given as 20 mg twice daily) to 60 mg/day (given either once daily or as 30 mg twice daily). For some patients, it may be desirable to start at 30 mg once daily for 1 week, to allow patients to adjust to CYMBALTA before increasing to 60 mg once daily. While a 120 mg/day dose was shown to be effective, there is no evidence that doses greater than 60 mg/day confer any additional benefits. Periodically reassess to determine the need for maintenance treatment and the appropriate dosage for such treatment.
2.3 Dosage for Treatment of Generalized Anxiety Disorder
Recommended Dosage in Adults Less than 65 Years of Age
For most adults less than 65 years of age with GAD, initiate CYMBALTA 60 mg once daily. For some patients, it may be desirable to start at 30 mg once daily for 1 week, to allow patients to adjust to CYMBALTA before increasing to 60 mg once daily. While a 120 mg once daily dosage was shown to be effective, there is no evidence that doses greater than 60 mg/day confer additional benefit. Nevertheless, if a decision is made to increase the dosage beyond 60 mg once daily, increase dosage in increments of 30 mg once daily. Periodically reassess to determine the continued need for maintenance treatment and the appropriate dosage for such treatment.
Recommended Dosage in Geriatric Patients
In geriatric patients with GAD, initiate CYMBALTA at a dosage of 30 mg once daily for 2 weeks before considering an increase to the target dose of 60 mg/day. Thereafter, patients may benefit from doses above 60 mg once daily. If a decision is made to increase the dose beyond 60 mg once daily, increase dose in increments of 30 mg once daily. The maximum dose studied was 120 mg per day.
Recommended Dosage in Pediatric Patients 7 to 17 Years of Age
Initiate CYMBALTA in pediatric patients 7 to 17 years of age with GAD at a dosage of 30 mg once daily for 2 weeks before considering an increase to 60 mg once daily. The recommended dosage range is 30 to 60 mg once daily. Some patients may benefit from dosages above 60 mg once daily. If a decision is made to increase the dose beyond 60 mg once daily, increase dosage in increments of 30 mg once daily. The maximum dose studied was 120 mg per day.
2.4 Dosage for Treatment of Diabetic Peripheral Neuropathic Pain in Adults
Administer 60 mg once daily in adults with diabetic peripheral neuropathic pain. There is no evidence that doses higher than 60 mg once daily confer additional significant benefit and the higher dosage is clearly less well tolerated. For patients for whom tolerability is a concern, a lower starting dose may be considered.
Since diabetes is frequently complicated by renal disease, consider a lower starting dosage and gradual increase in dosage for patients with renal impairment [see Dosage and Administration (2.7) and Use in Specific Populations (8.10)].
2.5 Dosage for Treatment of Fibromyalgia
Recommended Dosage in Adults
The recommended CYMBALTA dosage is 60 mg once daily in adults with fibromyalgia. Begin treatment at 30 mg once daily for 1 week, to allow patients to adjust to CYMBALTA before increasing to 60 mg once daily. Some patients may respond to the starting dosage. There is no evidence that dosages greater than 60 mg/day confer additional benefit, even in patients who do not respond to a 60 mg/day dosage, and higher dosages were associated with a higher rate of adverse reactions.
2.6 Dosage for Treatment of Chronic Musculoskeletal Pain in Adults
The recommended CYMBALTA dosage is 60 mg once daily in adults with chronic musculoskeletal pain. Begin treatment at 30 mg once daily for one week, to allow patients to adjust to CYMBALTA before increasing to 60 mg once daily. There is no evidence that higher dosages confer additional benefit, even in patients who do not respond to a 60 mg once daily dosage, and higher dosages are associated with a higher rate of adverse reactions [see Clinical Studies (14.6)].
2.7 Dosage in Patients with Hepatic Impairment or Severe Renal Impairment
Avoid use in patients with chronic liver disease or cirrhosis [see Warnings and Precautions (5.14) and Use in Specific Populations (8.9)].
Avoid use in patients with severe renal impairment, GFR <30 mL/minute [see Warnings and Precautions (5.14) and Use in Specific Populations (8.10)].
2.8 Discontinuing CYMBALTA
Adverse reactions after discontinuation of CYMBALTA, after abrupt or tapered discontinuation, include: dizziness, headache, nausea, diarrhea, paresthesia, irritability, vomiting, insomnia, anxiety, hyperhidrosis, and fatigue. A gradual reduction in dosage rather than abrupt cessation is recommended whenever possible [see Warnings and Precautions (5.7)].
2.9 Switching a Patient to or from a Monoamine Oxidase Inhibitor (MAOI) Intended to Treat Psychiatric Disorders
At least 14 days should elapse between discontinuation of an MAOI intended to treat psychiatric disorders and initiation of therapy with CYMBALTA. Conversely, at least 5 days should be allowed after stopping CYMBALTA before starting an MAOI intended to treat psychiatric disorders [see Contraindications (4)].
2.10 Use of CYMBALTA with Other MAOIs such as Linezolid or Methylene Blue
Do not start CYMBALTA in a patient who is being treated with linezolid or intravenous methylene blue because there is an increased risk of serotonin syndrome. In a patient who requires more urgent treatment of a psychiatric condition, other interventions, including hospitalization, should be considered [see Contraindications (4)].
In some cases, a patient already receiving CYMBALTA therapy may require urgent treatment with linezolid or intravenous methylene blue. If acceptable alternatives to linezolid or intravenous methylene blue treatment are not available and the potential benefits of linezolid or intravenous methylene blue treatment are judged to outweigh the risks of serotonin syndrome in a particular patient, CYMBALTA should be stopped promptly, and linezolid or intravenous methylene blue can be administered. The patient should be monitored for symptoms of serotonin syndrome for 5 days or until 24 hours after the last dose of linezolid or intravenous methylene blue, whichever comes first. Therapy with CYMBALTA may be resumed 24 hours after the last dose of linezolid or intravenous methylene blue [see Warnings and Precautions (5.4)].
The risk of administering methylene blue by non-intravenous routes (such as oral tablets or by local injection) or in intravenous doses much lower than 1 mg/kg with CYMBALTA is unclear. The clinician should, nevertheless, be aware of the possibility of emergent symptoms of serotonin syndrome with such use [see Warnings and Precautions (5.4)].
3 DOSAGE FORMS AND STRENGTHS
CYMBALTA is available as delayed-release capsules:
- 20 mg opaque green capsules imprinted with “Lilly 3235 20mg”
- 30 mg opaque white and blue capsules imprinted with “Lilly 3240 30mg”
- 60 mg opaque green and blue capsules imprinted with “Lilly 3270 60mg”
4 CONTRAINDICATIONS
The use of MAOIs intended to treat psychiatric disorders with CYMBALTA or within 5 days of stopping treatment with CYMBALTA is contraindicated because of an increased risk of serotonin syndrome. The use of CYMBALTA within 14 days of stopping an MAOI intended to treat psychiatric disorders is contraindicated [see Dosage and Administration (2.8) and Warnings and Precautions (5.4)].
Starting CYMBALTA in a patient who is being treated with MAOIs such as linezolid or intravenous methylene blue is also contraindicated because of an increased risk of serotonin syndrome [see Dosage and Administration (2.9) and Warnings and Precautions (5.4)].
5 WARNINGS AND PRECAUTIONS
5.1 Suicidal Thoughts and Behaviors in Children, Adolescents, and Young Adults
Patients with major depressive disorder (MDD), both adult and pediatric, may experience worsening of their depression and/or the emergence of suicidal ideation and behavior (suicidality) or unusual changes in behavior, whether or not they are taking antidepressant medications, and this risk may persist until significant remission occurs. Suicide is a known risk of depression and certain other psychiatric disorders, and these disorders themselves are the strongest predictors of suicide. There has been a long-standing concern, however, that antidepressants may have a role in inducing worsening of depression and the emergence of suicidality in certain patients during the early phases of treatment.
Pooled analyses of short-term placebo-controlled trials of antidepressant drugs (SSRIs and others) showed that these drugs increase the risk of suicidal thinking and behavior (suicidality) in children, adolescents, and young adults (ages 18-24) with major depressive disorder (MDD) and other psychiatric disorders. Short-term studies did not show an increase in the risk of suicidality with antidepressants compared to placebo in adults beyond age 24; there was a reduction with antidepressants compared to placebo in adults aged 65 and older.
The pooled analyses of placebo-controlled trials in children and adolescents with MDD, obsessive compulsive disorder (OCD), or other psychiatric disorders included a total of 24 short-term trials of 9 antidepressant drugs in over 4400 patients. The pooled analyses of placebo-controlled trials in adults with MDD or other psychiatric disorders included a total of 295 short-term trials (median duration of 2 months) of 11 antidepressant drugs in over 77,000 patients. There was considerable variation in risk of suicidality among drugs, but a tendency toward an increase in the younger patients for almost all drugs studied. There were differences in absolute risk of suicidality across the different indications, with the highest incidence in MDD. The risk of differences (drug vs placebo), however, were relatively stable within age strata and across indications. These risk differences (drug-placebo difference in the number of cases of suicidality per 1000 patients treated) are provided in Table 1.
| Age Range | Drug-Placebo Difference in Number of Cases of Suicidality per 1000 Patients Treated |
| Increases Compared to Placebo | |
| <18 | 14 additional cases |
| 18-24 | 5 additional cases |
| Decreases Compared to Placebo | |
| 25-64 | 1 fewer case |
| ≥65 | 6 fewer cases |
No suicides occurred in any of the pediatric CYMBALTA trials. There were suicides in the adult CYMBALTA trials, but the number was not sufficient to reach any conclusion about CYMBALTA effect on suicide.
It is unknown whether the suicidality risk extends to longer-term use, i.e., beyond several months. However, there is substantial evidence from placebo-controlled maintenance trials in adults with depression that the use of antidepressants can delay the recurrence of depression.
All patients being treated with antidepressants for any indication should be monitored appropriately and observed closely for clinical worsening, suicidality, and unusual changes in behavior, especially during the initial few months of a course of drug therapy, or at times of dose changes, either increases or decreases.
The following symptoms, anxiety, agitation, panic attacks, insomnia, irritability, hostility, aggressiveness, impulsivity, akathisia (psychomotor restlessness), hypomania, and mania, have been reported in adult and pediatric patients being treated with antidepressants for major depressive disorder as well as for other indications, both psychiatric and nonpsychiatric. Although a causal link between the emergence of such symptoms and either the worsening of depression and/or the emergence of suicidal impulses has not been established, there is concern that such symptoms may represent precursors to emerging suicidality.
Consideration should be given to changing the therapeutic regimen, including possibly discontinuing the medication, in patients whose depression is persistently worse, or who are experiencing emergent suicidality or symptoms that might be precursors to worsening depression or suicidality, especially if these symptoms are severe, abrupt in onset, or were not part of the patient's presenting symptoms.
If the decision has been made to discontinue treatment, medication should be tapered, as rapidly as is feasible, but with recognition that discontinuation can be associated with certain symptoms [see Dosage and Administration (2.8) and Warnings and Precautions (5.7)] for descriptions of the risks of discontinuation of CYMBALTA.
Families and caregivers of patients being treated with antidepressants for major depressive disorder or other indications, both psychiatric and nonpsychiatric, should be alerted about the need to monitor patients for the emergence of agitation, irritability, unusual changes in behavior, and the other symptoms described above, as well as the emergence of suicidality, and to report such symptoms immediately to health care providers. Such monitoring should include daily observation by families and caregivers. Prescriptions for CYMBALTA should be written for the smallest quantity of capsules consistent with good patient management, in order to reduce the risk of overdose.
Screening Patients for Bipolar Disorder
A major depressive episode may be the initial presentation of bipolar disorder. It is generally believed (though not established in controlled trials) that treating such an episode with an antidepressant alone may increase the likelihood of precipitation of a mixed/manic episode in patients at risk for bipolar disorder. Whether any of the symptoms described above represent such a conversion is unknown. However, prior to initiating treatment with an antidepressant, patients with depressive symptoms should be adequately screened to determine if they are at risk for bipolar disorder; such screening should include a detailed psychiatric history, including a family history of suicide, bipolar disorder, and depression. It should be noted that CYMBALTA is not approved for use in treating bipolar depression.
5.2 Hepatotoxicity
There have been reports of hepatic failure, sometimes fatal, in patients treated with CYMBALTA. These cases have presented as hepatitis with abdominal pain, hepatomegaly, and elevation of transaminase levels to more than twenty times the upper limit of normal (ULN) with or without jaundice, reflecting a mixed or hepatocellular pattern of liver injury. CYMBALTA should be discontinued in patients who develop jaundice or other evidence of clinically significant liver dysfunction and should not be resumed unless another cause can be established.
Cases of cholestatic jaundice with minimal elevation of transaminase levels have also been reported. Other postmarketing reports indicate that elevated transaminases, bilirubin, and alkaline phosphatase have occurred in patients with chronic liver disease or cirrhosis.
CYMBALTA increased the risk of elevation of serum transaminase levels in development program clinical trials. Liver transaminase elevations resulted in the discontinuation of 0.3% (92/34,756) of CYMBALTA-treated patients. In most patients, the median time to detection of the transaminase elevation was about two months. In adult placebo-controlled trials, for patients with normal and abnormal baseline ALT values, elevation of ALT >3 times the ULN occurred in 1.25% (144/11,496) of CYMBALTA-treated patients compared to 0.45% (39/8716) of placebo-treated patients. In adult placebo-controlled studies using a fixed dose design, there was evidence of a CYMBALTA dose response relationship for ALT and AST elevation of >3 times the ULN and >5 times the ULN, respectively.
Because it is possible that CYMBALTA and alcohol may interact to cause liver injury or that CYMBALTA may aggravate pre-existing liver disease, CYMBALTA should not be prescribed to patients with substantial alcohol use or evidence of chronic liver disease.
5.3 Orthostatic Hypotension, Falls and Syncope
Orthostatic hypotension, falls, and syncope have been reported in patients treated with the recommended CYMBALTA dosages. Syncope and orthostatic hypotension tend to occur within the first week of therapy but can occur at any time during CYMBALTA treatment, particularly after dose increases. The risk of falling appears to be related to the degree of orthostatic decrease in blood pressure (BP) as well as other factors that may increase the underlying risk of falls.
In an analysis of patients from all placebo-controlled trials, patients treated with CYMBALTA reported a higher rate of falls compared to patients treated with placebo. Risk appears to be related to the presence of orthostatic decrease in BP. The risk of BP decreases may be greater in patients taking concomitant medications that induce orthostatic hypotension (such as antihypertensives) or are potent CYP1A2 inhibitors [see Warnings and Precautions (5.12) and Drug Interactions (7.1)] and in patients taking CYMBALTA at doses above 60 mg daily. Consideration should be given to dose reduction or discontinuation of CYMBALTA in patients who experience symptomatic orthostatic hypotension, falls and/or syncope during CYMBALTA therapy.
Risk of falling also appeared to be proportional to a patient's underlying risk for falls and appeared to increase steadily with age. As geriatric patients tend to have a higher underlying risk for falls due to a higher prevalence of risk factors such as use of multiple medications, medical comorbidities and gait disturbances, the impact of increasing age by itself is unclear. Falls with serious consequences including fractures and hospitalizations have been reported with CYMBALTA use [see Adverse Reactions (6.1)].
5.4 Serotonin Syndrome
Serotonin-norepinephrine reuptake inhibitors (SNRIs), including CYMBALTA, can precipitate serotonin syndrome, a potentially life-threatening condition. The risk is increased with concomitant use of other serotonergic drugs (including triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, meperidine, methadone, tryptophan, buspirone, amphetamines, and St. John's Wort) and with drugs that impair metabolism of serotonin, i.e., MAOIs, [see Contraindications (4), Drug Interactions (7.13)]. Serotonin syndrome can also occur when these drugs are used alone.
Serotonin syndrome signs and symptoms may include mental status changes (e.g., agitation, hallucinations, delirium, and coma), autonomic instability (e.g., tachycardia, labile blood pressure, dizziness, diaphoresis, flushing, hyperthermia), neuromuscular symptoms (e.g., tremor, rigidity, myoclonus, hyperreflexia, incoordination), seizures, and gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea).
The concomitant use of CYMBALTA with MAOIs is contraindicated. In addition, do not initiate CYMBALTA in a patient being treated with MAOIs such as linezolid or intravenous methylene blue. No reports involved the administration of methylene blue by other routes (such as oral tablets or local tissue injection). If it is necessary to initiate treatment with an MAOI such as linezolid or intravenous methylene blue in a patient taking CYMBALTA, discontinue CYMBALTA before initiating treatment with the MAOI [see Contraindications (4) and Drug Interactions (7.13)].
Monitor all patients taking CYMBALTA for the emergence of serotonin syndrome. Discontinue treatment with CYMBALTA and any concomitant serotonergic agents immediately if the above symptoms occur, and initiate supportive symptomatic treatment. If concomitant use of CYMBALTA with other serotonergic drugs is clinically warranted, inform patients of the increased risk for serotonin syndrome and monitor for symptoms.
5.5 Increased Risk of Bleeding
Drugs that interfere with serotonin reuptake inhibition, including CYMBALTA, may increase the risk of bleeding events. Case reports and epidemiological studies (case-control and cohort design) have demonstrated an association between use of drugs that interfere with serotonin reuptake and the occurrence of gastrointestinal bleeding. A post-marketing study showed a higher incidence of postpartum hemorrhage in mothers taking CYMBALTA. Other bleeding events related to SSRI and SNRI use have ranged from ecchymoses, hematomas, epistaxis, and petechiae to life-threatening hemorrhages. Concomitant use of aspirin, nonsteroidal anti-inflammatory drugs (NSAIDs), warfarin, and other anti-coagulants may add to this risk.
Inform patients about the risk of increased bleeding associated with the concomitant use of CYMBALTA and NSAIDs, aspirin, or other drugs that affect coagulation [see Drug Interactions (7.4)].
5.6 Severe Skin Reactions
Severe skin reactions, including erythema multiforme and Stevens-Johnson Syndrome (SJS), can occur with CYMBALTA. The reporting rate of SJS associated with CYMBALTA use exceeds the general population background incidence rate for this serious skin reaction (1 to 2 cases per million person years). The reporting rate is generally accepted to be an underestimate due to underreporting.
CYMBALTA should be discontinued at the first appearance of blisters, peeling rash, mucosal erosions, or any other sign of hypersensitivity if no other etiology can be identified.
5.7 Discontinuation Syndrome
Discontinuation symptoms have been systematically evaluated in patients taking CYMBALTA. Following abrupt or tapered discontinuation in adult placebo-controlled clinical trials, the following symptoms occurred at 1% or greater and at a significantly higher rate in CYMBALTA-treated patients compared to those discontinuing from placebo: dizziness, headache, nausea, diarrhea, paresthesia, irritability, vomiting, insomnia, anxiety, hyperhidrosis, and fatigue.
During marketing of other SSRIs and SNRIs (serotonin and norepinephrine reuptake inhibitors), there have been spontaneous reports of adverse events occurring upon discontinuation of these drugs, particularly when abrupt, including the following: dysphoric mood, irritability, agitation, dizziness, sensory disturbances (e.g., paresthesias such as electric shock sensations), anxiety, confusion, headache, lethargy, emotional lability, insomnia, hypomania, tinnitus, and seizures. Although these events are generally self-limiting, some have been reported to be severe.
Patients should be monitored for these symptoms when discontinuing treatment with CYMBALTA. A gradual reduction in the dose rather than abrupt cessation is recommended whenever possible. If intolerable symptoms occur following a decrease in the dose or upon discontinuation of treatment, then resuming the previously prescribed dose may be considered. Subsequently, the healthcare provider may continue decreasing the dose but at a more gradual rate [see Dosage and Administration (2.8)].
5.8 Activation of Mania/Hypomania
In adult placebo-controlled trials in patients with MDD, activation of mania or hypomania was reported in 0.1% (4/3779) of CYMBALTA-treated patients and 0.04% (1/2536) of placebo-treated patients. No activation of mania or hypomania was reported in DPNP, GAD, fibromyalgia, or chronic musculoskeletal pain placebo-controlled trials. Activation of mania or hypomania has been reported in a small proportion of patients with mood disorders who were treated with other marketed drugs effective in the treatment of major depressive disorder. As with these other agents, CYMBALTA should be used cautiously in patients with a history of mania.
5.9 Angle-Closure Glaucoma
The pupillary dilation that occurs following use of many antidepressant drugs including CYMBALTA may trigger an angle closure attack in a patient with anatomically narrow angles who does not have a patent iridectomy.
5.10 Seizures
CYMBALTA has not been systematically evaluated in patients with a seizure disorder, and such patients were excluded from clinical studies. In adult placebo-controlled clinical trials, seizures/convulsions occurred in 0.02% (3/12,722) of patients treated with CYMBALTA and 0.01% (1/9513) of patients treated with placebo. CYMBALTA should be prescribed with care in patients with a history of a seizure disorder.
5.11 Increases in Blood Pressure
In adult placebo-controlled clinical trials across the approved adult populations from baseline to endpoint, CYMBALTA treatment was associated with mean increases of 0.5 mm Hg in systolic blood pressure and 0.8 mm Hg in diastolic blood pressure compared to mean decreases of 0.6 mm Hg systolic and 0.3 mm Hg diastolic in placebo-treated patients. There was no significant difference in the frequency of sustained (3 consecutive visits) elevated blood pressure. In a clinical pharmacology study designed to evaluate the effects of CYMBALTA on various parameters, including blood pressure at supratherapeutic doses with an accelerated dose titration, there was evidence of increases in supine blood pressure at doses up to 200 mg twice daily (approximately 3.3 times the maximum recommended dosage). At the highest 200 mg twice daily dose, the increase in mean pulse rate was 5.0 to 6.8 beats and increases in mean blood pressure were 4.7 to 6.8 mm Hg (systolic) and 4.5 to 7 mm Hg (diastolic) up to 12 hours after dosing.
Blood pressure should be measured prior to initiating treatment and periodically measured throughout treatment [see Adverse Reactions (6.1)].
5.12 Clinically Important Drug Interactions
Both CYP1A2 and CYP2D6 are responsible for CYMBALTA metabolism.
Potential for Other Drugs to Affect CYMBALTA
CYP1A2 Inhibitors — Co-administration of CYMBALTA with potent CYP1A2 inhibitors should be avoided [see Drug Interactions (7.1)].
CYP2D6 Inhibitors — Because CYP2D6 is involved in CYMBALTA metabolism, concomitant use of CYMBALTA with potent inhibitors of CYP2D6 would be expected to, and does, result in higher concentrations (on average of 60%) of CYMBALTA [see Drug Interactions (7.2)].
Potential for CYMBALTA to Affect Other Drugs
Drugs Metabolized by CYP2D6 — Co-administration of CYMBALTA with drugs that are extensively metabolized by CYP2D6 and that have a narrow therapeutic index, including certain antidepressants (tricyclic antidepressants [TCAs], such as nortriptyline, amitriptyline, and imipramine), phenothiazines and Type 1C antiarrhythmics (e.g., propafenone, flecainide), should be approached with caution. Plasma TCA concentrations may need to be monitored and the dose of the TCA may need to be reduced if a TCA is co-administered with CYMBALTA. Because of the risk of serious ventricular arrhythmias and sudden death potentially associated with elevated plasma levels of thioridazine, CYMBALTA and thioridazine should not be co-administered [see Drug Interactions (7.9)].
Other Clinically Important Drug Interactions
5.13 Hyponatremia
Hyponatremia may occur as a result of treatment with SSRIs and SNRIs, including CYMBALTA. In many cases, this hyponatremia appears to be the result of the syndrome of inappropriate antidiuretic hormone secretion (SIADH). Cases with serum sodium lower than 110 mmol/L have been reported with CYMBALTA use and appeared to be reversible when CYMBALTA was discontinued. Geriatric patients may be at greater risk of developing hyponatremia with SSRIs and SNRIs. Also, patients taking diuretics or who are otherwise volume depleted may be at greater risk [see Use in Specific Populations (8.5)]. Discontinuation of CYMBALTA should be considered in patients with symptomatic hyponatremia and appropriate medical intervention should be instituted.
Signs and symptoms of hyponatremia include headache, difficulty concentrating, memory impairment, confusion, weakness, and unsteadiness, which may lead to falls. More severe and/or acute cases have been associated with hallucination, syncope, seizure, coma, respiratory arrest, and death.
5.14 Use in Patients with Concomitant Illness
Clinical experience with CYMBALTA in patients with concomitant systemic illnesses is limited. There is no information on the effect that alterations in gastric motility may have on the stability of CYMBALTA's enteric coating. In extremely acidic conditions, CYMBALTA, unprotected by the enteric coating, may undergo hydrolysis to form naphthol. Caution is advised in using CYMBALTA in patients with conditions that may slow gastric emptying (e.g., some diabetics).
CYMBALTA has not been systematically evaluated in patients with a recent history of myocardial infarction or unstable coronary artery disease. Patients with these diagnoses were generally excluded from clinical studies during the product's premarketing testing.
Hepatic Impairment
Avoid use in patients with chronic liver disease or cirrhosis [see Dosage and Administration (2.7), Warnings and Precautions (5.2), and Use in Specific Populations (8.9)].
Severe Renal Impairment
Avoid use in patients with severe renal impairment, GFR <30 mL/minute. Increased plasma concentration of CYMBALTA, and especially of its metabolites, occurred in patients with end-stage renal disease (requiring dialysis) [see Dosage and Administration (2.7) and Use in Specific Populations (8.10)].
Glycemic Control in Patients with Diabetes
As observed in DPNP trials, CYMBALTA treatment worsened glycemic control in some patients with diabetes. In three clinical trials of CYMBALTA for the management of neuropathic pain associated with diabetic peripheral neuropathy [see Clinical Studies (14.4)], the mean duration of diabetes was approximately 12 years, the mean baseline fasting blood glucose was 176 mg/dL, and the mean baseline hemoglobin A1c (HbA1c) was 7.8%. In the 12-week acute treatment phase of these studies, CYMBALTA was associated with a small increase in mean fasting blood glucose as compared to placebo. In the extension phase of these studies, which lasted up to 52 weeks, mean fasting blood glucose increased by 12 mg/dL in the CYMBALTA group and decreased by 11.5 mg/dL in the routine care group. HbA1c increased by 0.5% in the CYMBALTA group and by 0.2% in the routine care group.
5.15 Urinary Hesitation and Retention
CYMBALTA is in a class of drugs known to affect urethral resistance. If symptoms of urinary hesitation develop during treatment with CYMBALTA, consideration should be given to the possibility that they might be drug-related.
In post marketing experience, cases of urinary retention have been observed. In some instances of urinary retention associated with CYMBALTA use, hospitalization and/or catheterization has been needed.
5.16 Sexual Dysfunction
Use of SNRIs, including CYMBALTA, may cause symptoms of sexual dysfunction [see Adverse Reactions (6.1)]. In male patients, SNRI use may result in ejaculatory delay or failure, decreased libido, and erectile dysfunction. In female patients, SNRI use may result in decreased libido and delayed or absent orgasm.
It is important for prescribers to inquire about sexual function prior to initiation of CYMBALTA and to inquire specifically about changes in sexual function during treatment, because sexual function may not be spontaneously reported. When evaluating changes in sexual function, obtaining a detailed history (including timing of symptom onset) is important because sexual symptoms may have other causes, including the underlying psychiatric disorder. Discuss potential management strategies to support patients in making informed decisions about treatment.
6 ADVERSE REACTIONS
The following serious adverse reactions are described below and elsewhere in the labeling:
- Suicidal Thoughts and Behaviors in Children, Adolescents, and Young Adults [see Boxed Warning and Warnings and Precautions (5.1)]
- Hepatotoxicity [see Warnings and Precautions (5.2)]
- Orthostatic Hypotension, Falls and Syncope [see Warnings and Precautions (5.3)]
- Serotonin Syndrome [see Warnings and Precautions (5.4)]
- Increased Risk of Bleeding [see Warnings and Precautions (5.5)]
- Severe Skin Reactions [see Warnings and Precautions (5.6)]
- Discontinuation Syndrome [see Warnings and Precautions (5.7)]
- Activation of Mania/Hypomania [see Warnings and Precautions (5.8)]
- Angle-Closure Glaucoma [see Warnings and Precautions (5.9)]
- Seizures [see Warnings and Precautions (5.10)]
- Increases in Blood Pressure [see Warnings and Precautions (5.11)]
- Clinically Important Drug Interactions [see Warnings and Precautions (5.12)]
- Hyponatremia [see Warnings and Precautions (5.13)]
- Urinary Hesitation and Retention [see Warnings and Precautions (5.15)]
- Sexual Dysfunction [see Warnings and Precautions (5.16)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The stated frequencies of adverse reactions represent the proportion of patients who experienced, at least once, one treatment-emergent adverse reaction of the type listed. A reaction was considered treatment-emergent if it occurred for the first time or worsened while receiving therapy following baseline evaluation.
Adverse Reactions in Adults
Adult Clinical Trial Database
The data described below reflect exposure to CYMBALTA in placebo-controlled adult trials for MDD (N=3779), GAD (N=1018), OA (N=503), CLBP (N=600), DPNP (N=906), and FM (N=1294). The age range in this pooled population was 17 to 89 years of age. In this pooled population, 66%, 61%, 61%, 43%, and 94% of adult patients were female; and 82%, 73%, 85%, 74%, and 86% of adult patients were Caucasian in the MDD, GAD, OA and CLBP, DPNP, and FM populations, respectively. Most patients received CYMBALTA dosages of a total of 60 to 120 mg per day [see Clinical Studies (14)]. The data below do not include results of the trial that evaluated the efficacy of CYMBALTA for the treatment of GAD in patients ≥65 years old (Study GAD-5) [see Clinical Studies (14.3)]; however, the adverse reactions observed in this geriatric population were generally similar to adverse reactions in the overall adult population.
Adverse Reactions Leading to Treatment Discontinuation in Adult Placebo-Controlled Trials
Major Depressive Disorder
Approximately 8.4% (319/3779) of CYMBALTA-treated patients in placebo-controlled adult trials for MDD discontinued treatment due to an adverse reaction, compared with 4.6% (117/2536) of placebo-treated patients. Nausea (CYMBALTA 1.1%, placebo 0.4%) was the only adverse reaction reported as a reason for discontinuation and considered to be drug-related (i.e., discontinuation occurring in at least 1% of the CYMBALTA-treated patients and at a rate of at least twice that of placebo-treated patients).
Generalized Anxiety Disorder
Approximately 13.7% (139/1018) of the CYMBALTA-treated patients in placebo-controlled adult trials for GAD discontinued treatment due to an adverse reaction, compared with 5% (38/767) for placebo-treated patients. Common adverse reactions reported as a reason for discontinuation and considered to be drug-related (as defined above) included nausea (CYMBALTA 3.3%, placebo 0.4%), and dizziness (CYMBALTA 1.3%, placebo 0.4%).
Diabetic Peripheral Neuropathic Pain
Approximately 12.9% (117/906) of the CYMBALTA-treated patients in placebo-controlled adult trials for DPNP discontinued treatment due to an adverse reaction, compared with 5.1% (23/448) for placebo-treated patients. Common adverse reactions reported as a reason for discontinuation and considered to be drug-related (as defined above) included nausea (CYMBALTA 3.5%, placebo 0.7%), dizziness (CYMBALTA 1.2%, placebo 0.4%), and somnolence (CYMBALTA 1.1%, placebo 0%).
Fibromyalgia
Approximately 17.5% (227/1294) of the CYMBALTA-treated patients in 3- to 6-month placebo-controlled adult trials for FM discontinued treatment due to an adverse reaction, compared with 10.1% (96/955) for placebo-treated patients. Adverse reactions reported as a reason for discontinuation and considered to be drug-related (as defined above) included nausea (CYMBALTA 2.0%, placebo 0.5%), headache (CYMBALTA 1.2%, placebo 0.3%), somnolence (CYMBALTA 1.1%, placebo 0%), and fatigue (CYMBALTA 1.1%, placebo 0.1%).
Chronic Pain due to Osteoarthritis
Approximately 15.7% (79/503) of the CYMBALTA-treated patients in 13-week, placebo-controlled adult trials for chronic pain due to OA discontinued treatment due to an adverse reaction, compared with 7.3% (37/508) for placebo-treated patients. Adverse reactions reported as a reason for discontinuation and considered to be drug-related (as defined above) included nausea (CYMBALTA 2.2%, placebo 1%).
Chronic Low Back Pain
Approximately 16.5% (99/600) of the CYMBALTA-treated patients in 13-week, placebo-controlled adult trials for CLBP discontinued treatment due to an adverse reaction, compared with 6.3% (28/441) for placebo-treated patients. Adverse reactions reported as a reason for discontinuation and considered to be drug-related (as defined above) included nausea (CYMBALTA 3%, placebo 0.7%), and somnolence (CYMBALTA 1%, placebo 0%).
Most Common Adverse Reactions in Adult Trials
The most commonly observed adverse reactions in CYMBALTA-treated patients (as defined above) were:
- Diabetic Peripheral Neuropathic Pain: nausea, somnolence, decreased appetite, constipation, hyperhidrosis, and dry mouth.
- Fibromyalgia: nausea, dry mouth, constipation, somnolence, decreased appetite, hyperhidrosis, and agitation.
- Chronic Pain due to Osteoarthritis: nausea, fatigue, constipation, dry mouth, insomnia, somnolence, and dizziness.
- Chronic Low Back Pain: nausea, dry mouth, insomnia, somnolence, constipation, dizziness, and fatigue.
The most commonly observed adverse reactions in CYMBALTA-treated patients in all the pooled adult populations (i.e., MDD, GAD, DPNP, FM, OA, and CLBP) (incidence of at least 5% and at least twice the incidence in placebo-treated patients) were nausea, dry mouth, somnolence, constipation, decreased appetite, and hyperhidrosis.
Table 2 displays the incidence of adverse reactions in placebo-controlled trials for approved adult populations (i.e., MDD, GAD, DPNP, FM, OA, and CLBP) that occurred in 5% or more of CYMBALTA-treated patients and with an incidence greater than placebo-treated patients.
|
a Includes adults with MDD, GAD, DPNP, FM, and chronic musculoskeletal pain. The inclusion of an event in the table is determined based on the percentages before rounding; however, the percentages displayed in the table are rounded to the nearest integer. |
||
|
b Also includes asthenia. |
||
|
c Events for which there was a significant dose-dependent relationship in fixed-dose studies, excluding three MDD studies which did not have a placebo lead-in period or dose titration. |
||
|
d Also includes initial insomnia, middle insomnia, and early morning awakening. |
||
|
e Also includes hypersomnia and sedation. |
||
|
f Also includes abdominal discomfort, abdominal pain lower, abdominal pain upper, abdominal tenderness, and gastrointestinal pain. |
||
Adverse Reaction |
Percentage of Patients Reporting Reaction | |
| CYMBALTA (N=8100) |
Placebo (N=5655) |
|
| Nauseac | 23 | 8 |
| Headache | 14 | 12 |
| Dry mouth | 13 | 5 |
| Somnolencee | 10 | 3 |
| Fatigueb,c | 9 | 5 |
| Insomniad | 9 | 5 |
| Constipationc | 9 | 4 |
| Dizzinessc | 9 | 5 |
| Diarrhea | 9 | 6 |
| Decreased appetitec | 7 | 2 |
| Hyperhidrosisc | 6 | 1 |
| Abdominal painf | 5 | 4 |
Adverse Reactions in Pooled MDD and GAD Trials in Adults
Table 3 displays the incidence of adverse reactions in MDD and GAD placebo-controlled adult trials that occurred in 2% or more of CYMBALTA-treated patients and with an incidence greater than placebo-treated patients.
|
a The inclusion of an event in the table is determined based on the percentages before rounding; however, the percentages displayed in the table are rounded to the nearest integer. |
||
|
b For GAD, there were no adverse reactions that were significantly different between treatments in adults ≥65 years that were also not significant in the adults <65 years. |
||
|
c Events for which there was a significant dose-dependent relationship in fixed-dose studies, excluding three MDD studies which did not have a placebo lead-in period or dose titration. |
||
|
d Includes abdominal pain upper, abdominal pain lower, abdominal tenderness, abdominal discomfort, and gastrointestinal pain. |
||
|
e Includes asthenia. |
||
|
f Includes hypersomnia and sedation. |
||
|
g Includes initial insomnia, middle insomnia, and early morning awakening. |
||
|
h Includes feeling jittery, nervousness, restlessness, tension and psychomotor hyperactivity. |
||
|
i Includes loss of libido. |
||
|
j Includes anorgasmia. |
||
| System Organ Class / Adverse Reaction | Percentage of Patients Reporting Reaction | |
| CYMBALTA (N=4797) |
Placebo (N=3303) |
|
| Cardiac Disorders | ||
| Palpitations | 2 | 1 |
| Eye Disorders | ||
| Vision blurred | 3 | 1 |
| Gastrointestinal Disorders | ||
| Nauseac | 23 | 8 |
| Dry mouth | 14 | 6 |
| Constipationc | 9 | 4 |
| Diarrhea | 9 | 6 |
| Abdominal paind | 5 | 4 |
| Vomiting | 4 | 2 |
| General Disorders and Administration Site Conditions | ||
| Fatiguee | 9 | 5 |
| Metabolism and Nutrition Disorders | ||
| Decreased appetitec | 6 | 2 |
| Nervous System Disorders | ||
| Headache | 14 | 14 |
| Dizzinessc | 9 | 5 |
| Somnolencef | 9 | 3 |
| Tremor | 3 | 1 |
| Psychiatric Disorders | ||
| Insomniag | 9 | 5 |
| Agitationh | 4 | 2 |
| Anxiety | 3 | 2 |
| Reproductive System and Breast Disorders | ||
| Erectile dysfunction | 4 | 1 |
| Ejaculation delayedc | 2 | 1 |
| Libido decreasedi | 3 | 1 |
| Orgasm abnormalj | 2 | <1 |
| Respiratory, Thoracic, and Mediastinal Disorders | ||
| Yawning | 2 | <1 |
| Skin and Subcutaneous Tissue Disorders | ||
| Hyperhidrosis | 6 | 2 |
Adverse Reactions in the DPNP, FM, OA, and CLBP Adult Trials
Table 4 displays the incidence of adverse reactions that occurred in 2% or more of CYMBALTA-treated patients (determined prior to rounding) in the premarketing acute phase of DPNP, FM, OA, and CLBP placebo-controlled adult trials and with an incidence greater than placebo-treated patients.
|
a The inclusion of an event in the table is determined based on the percentages before rounding; however, the percentages displayed in the table are rounded to the nearest integer. |
||
|
b Incidence of 120 mg/day is significantly greater than the incidence for 60 mg/day. |
||
|
c Includes abdominal discomfort, lower abdominal pain, upper abdominal pain, abdominal tenderness and gastrointestinal pain. |
||
|
d Includes asthenia. |
||
|
e Includes myalgia and neck pain. |
||
|
f Includes hypersomnia and sedation. |
||
|
g Includes hypoaesthesia, facial hypoaesthesia, genital hypoaesthesia and oral paraesthesia. |
||
|
h Includes initial insomnia, middle insomnia, and early morning awakening. |
||
|
i Includes feeling jittery, nervousness, restlessness, tension and psychomotor hyperactivity. |
||
|
j Includes ejaculation failure. |
||
|
k Includes hot flush. |
||
|
l Includes increased diastolic blood pressure, increased systolic blood pressure, diastolic hypertension, essential hypertension, hypertension, hypertensive crisis, labile hypertension, orthostatic hypertension, secondary hypertension, and systolic hypertension. |
||
| System Organ Class / Adverse Reaction | Percentage of Patients Reporting Reaction | |
| CYMBALTA (N=3303) |
Placebo (N=2352) |
|
| Gastrointestinal Disorders | ||
| Nausea | 23 | 7 |
| Dry Mouthb | 11 | 3 |
| Constipationb | 10 | 3 |
| Diarrhea | 9 | 5 |
| Abdominal Painc | 5 | 4 |
| Vomiting | 3 | 2 |
| Dyspepsia | 2 | 1 |
| General Disorders and Administration Site Conditions | ||
| Fatigued | 11 | 5 |
| Infections and Infestations | ||
| Nasopharyngitis | 4 | 4 |
| Upper Respiratory Tract Infection | 3 | 3 |
| Influenza | 2 | 2 |
| Metabolism and Nutrition Disorders | ||
| Decreased Appetiteb | 8 | 1 |
| Musculoskeletal and Connective Tissue | ||
| Musculoskeletal Paine | 3 | 3 |
| Muscle Spasms | 2 | 2 |
| Nervous System Disorders | ||
| Headache | 13 | 8 |
| Somnolenceb,f | 11 | 3 |
| Dizziness | 9 | 5 |
| Paraesthesiag | 2 | 2 |
| Tremorb | 2 | <1 |
| Psychiatric Disorders | ||
| Insomniab,h | 10 | 5 |
| Agitationi | 3 | 1 |
| Reproductive System and Breast Disorders | ||
| Erectile Dysfunctionb | 4 | <1 |
| Ejaculation Disorderj | 2 | <1 |
| Respiratory, Thoracic, and Mediastinal Disorders | ||
| Cough | 2 | 2 |
| Skin and Subcutaneous Tissue Disorders | ||
| Hyperhidrosis | 6 | 1 |
| Vascular Disorders | ||
| Flushingk | 3 | 1 |
| Blood pressure increasedl | 2 | 1 |
Effects on Male and Female Sexual Function in Adults with MDD
Changes in sexual desire, sexual performance and sexual satisfaction often occur as manifestations of psychiatric disorders or diabetes, but they may also be a consequence of pharmacologic treatment. Because adverse sexual reactions are presumed to be voluntarily underreported, the Arizona Sexual Experience Scale (ASEX), a validated measure designed to identify sexual adverse reactions, was used prospectively in 4 MDD placebo-controlled adult trials (Studies MDD-1, MDD-2, MDD-3, and MDD-4) [see Clinical Studies (14.2)]. The ASEX scale includes five questions that pertain to the following aspects of sexual function: 1) sex drive, 2) ease of arousal, 3) ability to achieve erection (men) or lubrication (women), 4) ease of reaching orgasm, and 5) orgasm satisfaction. Positive numbers signify a worsening of sexual function from baseline. Negative numbers signify an improvement from a baseline level of dysfunction, which is commonly seen in depressed patients.
In these trials, CYMBALTA-treated male patients experienced significantly more sexual dysfunction, as measured by the total score on the ASEX and the ability to reach orgasm, than placebo-treated male patients (see Table 5). CYMBALTA-treated female patients did not experience more sexual dysfunction than placebo-treated female patients as measured by ASEX total score. Healthcare providers should routinely inquire about possible sexual adverse reactions in CYMBALTA-treated patients.
|
a n=Number of patients with non-missing change score for ASEX total. |
||||
|
b p=0.013 versus placebo. |
||||
|
c p<0.001 versus placebo. |
||||
| Male Patientsa | Female Patientsa | |||
| CYMBALTA (n=175) |
Placebo (n=83) |
CYMBALTA (n=241) |
Placebo (n=126) |
|
| ASEX Total (Items 1-5) | 0.56b | -1.07 | -1.15 | -1.07 |
| Item 1 — Sex drive | -0.07 | -0.12 | -0.32 | -0.24 |
| Item 2 — Arousal | 0.01 | -0.26 | -0.21 | -0.18 |
| Item 3 — Ability to achieve erection (men); Lubrication (women) | 0.03 | -0.25 | -0.17 | -0.18 |
| Item 4 — Ease of reaching orgasm | 0.40c | -0.24 | -0.09 | -0.13 |
| Item 5 — Orgasm satisfaction | 0.09 | -0.13 | -0.11 | -0.17 |
Vital Sign Changes in Adults
In placebo-controlled clinical trials across approved adult populations for change from baseline to endpoint, CYMBALTA-treated patients had mean increases of 0.23 mm Hg in systolic blood pressure (SBP) and 0.73 mm Hg in diastolic blood pressure (DBP) compared to mean decreases of 1.09 mm Hg in SBP and 0.55 mm Hg in DBP in placebo-treated patients. There was no significant difference in the frequency of sustained (3 consecutive visits) elevated blood pressure [see Warnings and Precautions (5.3, 5.11)].
CYMBALTA treatment, for up to 26 weeks in placebo-controlled trials across approved adult populations, typically caused a small increase in heart rate for change from baseline to endpoint compared to placebo of up to 1.37 beats per minute (increase of 1.20 beats per minute in CYMBALTA-treated patients, decrease of 0.17 beats per minute in placebo-treated patients).
Laboratory Changes in Adults
CYMBALTA treatment in placebo-controlled clinical trials across approved adult populations, was associated with small mean increases from baseline to endpoint in ALT, AST, CPK, and alkaline phosphatase; infrequent, modest, transient, abnormal values were observed for these analytes in CYMBALTA-treated patients when compared with placebo-treated patients [see Warnings and Precautions (5.2)]. High bicarbonate, cholesterol, and abnormal (high or low) potassium, were observed more frequently in CYMBALTA-treated patients compared to placebo-treated patients.
Other Adverse Reactions Observed During the Clinical Trial Evaluation of CYMBALTA in Adults
Following is a list of adverse reactions reported by patients treated with CYMBALTA in clinical adult trials. In clinical trials of all approved adult populations, 34,756 patients were treated with CYMBALTA. Of these, 27% (9337) took CYMBALTA for at least 6 months, and 12% (4317) took CYMBALTA for at least one year. The following listing is not intended to include reactions (1) already listed in previous tables or elsewhere in labeling, (2) for which a drug cause was remote, (3) which were so general as to be uninformative, (4) which were not considered to have significant clinical implications, or (5) which occurred at a rate equal to or less than placebo.
Reactions are categorized by body system according to the following definitions: frequent adverse reactions are those occurring in at least 1/100 patients; infrequent adverse reactions are those occurring in 1/100 to 1/1000 patients; rare reactions are those occurring in fewer than 1/1000 patients.
- Cardiac Disorders — Frequent: palpitations; Infrequent: myocardial infarction, tachycardia, and Takotsubo cardiomyopathy.
- Ear and Labyrinth Disorders — Frequent: vertigo; Infrequent: ear pain and tinnitus.
- Endocrine Disorders — Infrequent: hypothyroidism.
- Eye Disorders — Frequent: vision blurred; Infrequent: diplopia, dry eye, and visual impairment.
- Gastrointestinal Disorders — Frequent: flatulence; Infrequent: dysphagia, eructation, gastritis, gastrointestinal hemorrhage, halitosis, and stomatitis; Rare: gastric ulcer.
- General Disorders and Administration Site Conditions — Frequent: chills/rigors; Infrequent: falls, feeling abnormal, feeling hot and/or cold, malaise, and thirst; Rare: gait disturbance.
- Infections and Infestations — Infrequent: gastroenteritis and laryngitis.
- Investigations — Frequent: weight increased, weight decreased; Infrequent: blood cholesterol increased.
- Metabolism and Nutrition Disorders — Infrequent: dehydration and hyperlipidemia; Rare: dyslipidemia.
- Musculoskeletal and Connective Tissue Disorders — Frequent: musculoskeletal pain; Infrequent: muscle tightness and muscle twitching.
- Nervous System Disorders — Frequent: dysgeusia, lethargy, and paraesthesia/hypoesthesia; Infrequent: disturbance in attention, dyskinesia, myoclonus, and poor quality sleep; Rare: dysarthria.
- Psychiatric Disorders — Frequent: abnormal dreams and sleep disorder; Infrequent: apathy, bruxism, disorientation/confusional state, irritability, mood swings, and suicide attempt; Rare: completed suicide.
- Renal and Urinary Disorders — Frequent: urinary frequency; Infrequent: dysuria, micturition urgency, nocturia, polyuria, and urine odor abnormal.
- Reproductive System and Breast Disorders — Frequent: anorgasmia/orgasm abnormal; Infrequent: menopausal symptoms, sexual dysfunction, and testicular pain; Rare: menstrual disorder.
- Respiratory, Thoracic and Mediastinal Disorders — Frequent: yawning, oropharyngeal pain; Infrequent: throat tightness.
- Skin and Subcutaneous Tissue Disorders — Frequent: pruritus; Infrequent: cold sweat, dermatitis contact, erythema, increased tendency to bruise, night sweats, and photosensitivity reaction; Rare: ecchymosis.
- Vascular Disorders — Frequent: hot flush; Infrequent: flushing, orthostatic hypotension, and peripheral coldness.
Adverse Reactions Observed in Placebo-Controlled Clinical Trials in Pediatric Patients
Pediatric Clinical Trial Database
The data described below reflect exposure to CYMBALTA (N=567) in pediatric patients aged 7 to 18 years of age from two 10-week, placebo-controlled trials in patients with MDD (N=341) (Studies MDD-6 and MDD-7), one 10-week placebo-controlled trial in GAD (N=135) (Study GAD-6), and a 13-week trial in fibromyalgia (N=91). CYMBALTA is not approved for the treatment of MDD in pediatric patients [see Use in Specific Populations (8.4)]. Of the CYMBALTA-treated patients in these studies, 36% were 7 to 11 years of age (64% were between 12 to 18 years old), 55% were female, and 69% were Caucasian. Patients received 30 to 120 mg of CYMBALTA per day during placebo-controlled acute treatment studies. In the pediatric MDD, GAD, and fibromyalgia trials up to 40 weeks long, there were 988 CYMBALTA-treated pediatric patients aged 7 to 17 years of age (most patients received 30-120 mg per day) – 35% were 7 to 11 years of age (65% were 12 to 17 years old) and 56% were female.
Most Common Adverse Reactions in Pediatric Trials
The most common adverse reactions (≥5% in CYMBALTA-treated patients and at least twice the incidence of placebo-treated patients) in all pooled pediatric populations (MDD, GAD, and fibromyalgia) were decreased weight, decreased appetite, nausea, vomiting, fatigue, and diarrhea.
Adverse Reactions in Pediatric Patients Aged 7 to 17 Years Old with MDD and GAD
The adverse reaction profile observed in clinical trials in pediatric patients aged 7 to 18 years old with MDD and GAD was consistent with the adverse reaction profile observed in adult clinical trials. The most common (≥5% and twice placebo) adverse reactions observed in these pediatric clinical trials included: nausea, diarrhea, decreased weight, and dizziness.
Table 6 provides the incidence of adverse reactions in MDD and GAD pediatric placebo-controlled trials that occurred in greater than 2% of patients treated with CYMBALTA and with an incidence greater than patients treated with placebo. CYMBALTA is not approved in the treatment of MDD in pediatric patients [see Use in Specific Populations (8.4)].
|
a CYMBALTA is not approved for the treatment of pediatric MDD [see Use in Specific Populations (8.4)]. The inclusion of an event in the table is determined based on the percentages before rounding; however, the percentages displayed in the table are rounded to the nearest integer. |
||
|
b Also includes abdominal pain upper, abdominal pain lower, abdominal tenderness, abdominal discomfort, and gastrointestinal pain. |
||
|
c Also includes asthenia. |
||
|
d Frequency based on weight measurement meeting potentially clinically significant threshold of ≥3.5% weight loss (N=467 CYMBALTA; N=354 Placebo). |
||
|
e Also includes hypersomnia and sedation. |
||
|
f Also includes initial insomnia, insomnia, middle insomnia, and terminal insomnia. |
||
| System Organ Class/Adverse Reaction | Percentage of Pediatric Patients Reporting Reaction | |
| CYMBALTA (N=476) |
Placebo (N=362) |
|
| Gastrointestinal Disorders | ||
| Nausea | 18 | 8 |
| Abdominal Painb | 13 | 10 |
| Vomiting | 9 | 4 |
| Diarrhea | 6 | 3 |
| Dry Mouth | 2 | 1 |
| General Disorders and Administration Site Conditions | ||
| Fatiguec | 7 | 5 |
| Investigations | ||
| Decreased Weightd | 14 | 6 |
| Metabolism and Nutrition Disorders | ||
| Decreased Appetite | 10 | 5 |
| Nervous System Disorders | ||
| Headache | 18 | 13 |
| Somnolencee | 11 | 6 |
| Dizziness | 8 | 4 |
| Psychiatric Disorders | ||
| Insomniaf | 7 | 4 |
| Respiratory, Thoracic, and Mediastinal Disorders | ||
| Oropharyngeal Pain | 4 | 2 |
| Cough | 3 | 1 |
Other adverse reactions that occurred at an incidence of less than 2% and were reported by more CYMBALTA-treated patients than placebo-treated patients in pediatric MDD and GAD clinical trials included: abnormal dreams (including nightmare), anxiety, flushing (including hot flush), hyperhidrosis, palpitations, pulse increased, and tremor (CYMBALTA is not approved to treat pediatric patients with MDD).
The most commonly reported symptoms following discontinuation of CYMBALTA in pediatric MDD and GAD clinical trials included headache, dizziness, insomnia, and abdominal pain [see Warnings and Precautions (5.7)].
Growth (Height and Weight) in Pediatric Patients 7 to 17 Years Old with GAD and MDD
Decreased appetite and weight loss have been observed in association with the use of SSRIs and SNRIs. CYMBALTA-treated pediatric patients in clinical trials experienced a 0.1 kg mean decrease in weight at 10 weeks, compared with a mean weight gain of approximately 0.9 kg in placebo-treated pediatric patients. The proportion of patients who experienced a clinically significant decrease in weight (≥3.5%) was greater in the CYMBALTA group than in the placebo group (16% and 6%, respectively). Subsequently, over the 4- to 6-month uncontrolled extension periods, CYMBALTA-treated patients on average trended toward recovery to their expected baseline weight percentile based on population data from age- and sex-matched peers.
In studies up to 9 months, CYMBALTA-treated pediatric patients experienced an increase in height of 1.7 cm on average (2.2 cm increase in patients 7 to 11 years of age and 1.3 cm increase in patients 12 to 17 years of age). While height increase was observed during these studies, a mean decrease of 1% in height percentile was observed (decrease of 2% in patients 7 to 11 years of age and increase of 0.3% in patients 12 to 17 years of age). Weight and height should be monitored regularly in pediatric patients treated with CYMBALTA [see Use in Specific Populations (8.4)].
Adverse Reactions in Pediatric Patients Aged 13 to 17 Years Old with Fibromyalgia
Table 7 provides the incidence of adverse reactions in a fibromyalgia pediatric placebo-controlled trial (Study FM-4) that occurred in greater than 5% of patients treated with CYMBALTA and with an incidence greater than patients treated with placebo [see Clinical Studies (14.5)].
|
a The inclusion of an event in the table is determined based on the percentages before rounding; however, the percentages displayed in the table are rounded to the nearest integer. |
||
|
b Frequency based on weight measurement meeting potentially clinically significant threshold of ≥3.5% weight loss (N=89 CYMBALTA; N=92 Placebo). |
||
| CYMBALTA (N=91) |
Placebo (N=93) |
|
| Nausea | 25% | 15% |
| Decreased appetite | 15% | 3% |
| Vomiting | 15% | 5% |
| Decreased weightb | 15% | 5% |
| Headache | 14% | 11% |
| Nasopharyngitis | 9% | 2% |
| Somnolence | 9% | 3% |
| Upper respiratory tract infection | 7% | 2% |
| Viral gastroenteritis | 5% | 0% |
| Fatigue | 5% | 2% |
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of CYMBALTA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Adverse reactions reported since market introduction that were temporally related to CYMBALTA therapy and not mentioned elsewhere in labeling include: acute pancreatitis, anaphylactic reaction, aggression and anger (particularly early in treatment or after treatment discontinuation), angioneurotic edema, angle-closure glaucoma, colitis (microscopic or unspecified), cutaneous vasculitis (sometimes associated with systemic involvement), extrapyramidal disorder, galactorrhea, gynecological bleeding, hallucinations, hyperglycemia, hyperprolactinemia, hypersensitivity, hypertensive crisis, muscle spasm, rash, restless legs syndrome, seizures upon treatment discontinuation, supraventricular arrhythmia, tinnitus (upon treatment discontinuation), trismus, and urticaria.
7 DRUG INTERACTIONS
Both CYP1A2 and CYP2D6 are responsible for duloxetine metabolism.
7.1 Inhibitors of CYP1A2
When CYMBALTA 60 mg was co-administered with fluvoxamine 100 mg, a potent CYP1A2 inhibitor, to male subjects (n=14) duloxetine AUC was increased approximately 6-fold, the Cmax was increased about 2.5-fold, and duloxetine t1/2 was increased approximately 3-fold. Other drugs that inhibit CYP1A2 metabolism include cimetidine and quinolone antimicrobials such as ciprofloxacin and enoxacin [see Warnings and Precautions (5.12)].
7.2 Inhibitors of CYP2D6
Concomitant use of CYMBALTA (40 mg once daily) with paroxetine (20 mg once daily) increased the concentration of duloxetine AUC by about 60%, and greater degrees of inhibition are expected with higher doses of paroxetine. Similar effects would be expected with other potent CYP2D6 inhibitors (e.g., fluoxetine, quinidine) [see Warnings and Precautions (5.12)].
7.3 Dual Inhibition of CYP1A2 and CYP2D6
Concomitant administration of CYMBALTA 40 mg twice daily with fluvoxamine 100 mg, a potent CYP1A2 inhibitor, to CYP2D6 poor metabolizer subjects (n=14) resulted in a 6-fold increase in duloxetine AUC and Cmax.
7.4 Drugs that Interfere with Hemostasis (e.g., NSAIDs, Aspirin, and Warfarin)
Serotonin release by platelets plays an important role in hemostasis. Epidemiological studies of the case-control and cohort design that have demonstrated an association between use of psychotropic drugs that interfere with serotonin reuptake and the occurrence of upper gastrointestinal bleeding have also shown that concurrent use of an NSAID or aspirin may potentiate this risk of bleeding. Altered anticoagulant effects, including increased bleeding, have been reported when SSRIs or SNRIs are co-administered with warfarin. Concomitant administration of warfarin (2-9 mg once daily) under steady state conditions with CYMBALTA 60 or 120 mg once daily for up to 14 days in healthy subjects (n=15) did not significantly change INR from baseline (mean INR changes ranged from 0.05 to +0.07). The total warfarin (protein bound plus free drug) pharmacokinetics (AUCτ,ss, Cmax,ss or tmax,ss) for both R- and S-warfarin were not altered by duloxetine. Because of the potential effect of duloxetine on platelets, patients receiving warfarin therapy should be carefully monitored when CYMBALTA is initiated or discontinued [see Warnings and Precautions (5.5)].
7.5 Lorazepam
Under steady-state conditions for CYMBALTA (60 mg Q 12 hours) and lorazepam (2 mg Q 12 hours), the pharmacokinetics of duloxetine were not affected by co-administration.
7.6 Temazepam
Under steady-state conditions for CYMBALTA (20 mg qhs) and temazepam (30 mg qhs), the pharmacokinetics of duloxetine were not affected by co-administration.
7.7 Drugs that Affect Gastric Acidity
CYMBALTA has an enteric coating that resists dissolution until reaching a segment of the gastrointestinal tract where the pH exceeds 5.5. In extremely acidic conditions, CYMBALTA, unprotected by the enteric coating, may undergo hydrolysis to form naphthol. Caution is advised in using CYMBALTA in patients with conditions that may slow gastric emptying (e.g., some diabetics). Drugs that raise the gastrointestinal pH may lead to an earlier release of duloxetine. However, co-administration of CYMBALTA with aluminum- and magnesium-containing antacids (51 mEq) or CYMBALTA with famotidine, had no significant effect on the rate or extent of duloxetine absorption after administration of a 40 mg oral dose. It is unknown whether the concomitant administration of proton pump inhibitors affects duloxetine absorption [see Warnings and Precautions (5.14)].
7.8 Drugs Metabolized by CYP1A2
In vitro drug interaction studies demonstrate that duloxetine does not induce CYP1A2 activity. Therefore, an increase in the metabolism of CYP1A2 substrates (e.g., theophylline, caffeine) resulting from induction is not anticipated, although clinical studies of induction have not been performed. Duloxetine is an inhibitor of the CYP1A2 isoform in in vitro studies, and in two clinical studies the average (90% confidence interval) increase in theophylline AUC was 7% (1%-15%) and 20% (13%-27%) when co-administered with CYMBALTA (60 mg twice daily).
7.9 Drugs Metabolized by CYP2D6
Duloxetine is a moderate inhibitor of CYP2D6. When CYMBALTA was administered (at a dose of 60 mg twice daily) in conjunction with a single 50 mg dose of desipramine, a CYP2D6 substrate, the AUC of desipramine increased 3-fold [see Warnings and Precautions (5.12)].
7.10 Drugs Metabolized by CYP2C9
Results of in vitro studies demonstrate that duloxetine does not inhibit activity. In a clinical study, the pharmacokinetics of S-warfarin, a CYP2C9 substrate, were not significantly affected by duloxetine [see Drug Interactions (7.4)].
7.11 Drugs Metabolized by CYP3A
Results of in vitro studies demonstrate that duloxetine does not inhibit or induce CYP3A activity. Therefore, an increase or decrease in the metabolism of CYP3A substrates (e.g., oral contraceptives and other steroidal agents) resulting from induction or inhibition is not anticipated, although clinical studies have not been performed.
7.12 Drugs Metabolized by CYP2C19
Results of in vitro studies demonstrate that duloxetine does not inhibit CYP2C19 activity at therapeutic concentrations. Inhibition of the metabolism of CYP2C19 substrates is therefore not anticipated, although clinical studies have not been performed.
7.13 Monoamine Oxidase Inhibitors (MAOIs)
[See Dosage and Administration (2.9, 2.10), Contraindications (4), and Warnings and Precautions (5.4)].
7.14 Other Serotonergic Drugs
The concomitant use of serotonergic drugs (including other SNRIs, SSRIs, triptans, tricyclic antidepressants, opioids, lithium, buspirone, amphetamines, tryptophan, and St. John's Wort) with CYMBALTA increases the risk of serotonin syndrome. Monitor patients for signs and symptoms of serotonin syndrome, particularly during treatment initiation and dosage increases. If serotonin syndrome occurs, consider discontinuation of CYMBALTA and/or concomitant serotonergic drugs [see Warnings and Precautions (5.4)].
7.15 Alcohol
When CYMBALTA and ethanol were administered several hours apart so that peak concentrations of each would coincide, CYMBALTA did not increase the impairment of mental and motor skills caused by alcohol.
In the CYMBALTA clinical trials database, three CYMBALTA-treated patients had liver injury as manifested by ALT and total bilirubin elevations, with evidence of obstruction. Substantial intercurrent ethanol use was present in each of these cases, and this may have contributed to the abnormalities seen [see Warnings and Precautions (5.2, 5.12)].
7.17 Drugs Highly Bound to Plasma Protein
Because duloxetine is highly bound to plasma protein, administration of CYMBALTA to a patient taking another drug that is highly protein bound may cause increased free concentrations of the other drug, potentially resulting in adverse reactions. However, co-administration of CYMBALTA (60 or 120 mg) with warfarin (2-9 mg), a highly protein-bound drug, did not result in significant changes in INR and in the pharmacokinetics of either total S-or total R-warfarin (protein bound plus free drug) [see Drug Interactions (7.4)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors the pregnancy outcomes in women exposed to antidepressants, including CYMBALTA, during pregnancy. Healthcare providers are encouraged to register patients by contacting the National Pregnancy Registry for Antidepressants at 1-866-961-2388 or online at
https://womensmentalhealth.org/research/pregnancyregistry/.
Risk Summary
Data from a postmarketing retrospective cohort study indicate that use of duloxetine in the month before delivery may be associated with an increased risk of postpartum hemorrhage. Data from published literature and from a postmarketing retrospective cohort study have not identified a clear drug-associated risk of major birth defects or other adverse developmental outcomes (see Data). There are risks associated with untreated depression and fibromyalgia in pregnancy, and with exposure to SNRIs and SSRIs, including CYMBALTA, during pregnancy (see Clinical Considerations).
In rats and rabbits treated with duloxetine during the period of organogenesis, fetal weights were decreased but there was no evidence of developmental effects at doses up to 3 and 6 times, respectively, the maximum recommended human dose (MRHD) of 120 mg/day given to adolescents on a mg/m2 basis. When duloxetine was administered orally to pregnant rats throughout gestation and lactation, pup weights at birth and pup survival to 1 day postpartum were decreased at a dose 2 times the MRHD given to adolescents on a mg/m2 basis. At this dose, pup behaviors consistent with increased reactivity, such as increased startle response to noise and decreased habituation of locomotor activity were observed. Post-weaning growth was not adversely affected.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Disease-associated Maternal and/or Embryo/Fetal Risk
Women who discontinue antidepressants during pregnancy are more likely to experience a relapse of major depression than women who continue antidepressants. This finding is from a prospective, longitudinal study that followed 201 pregnant women with a history of major depressive disorder who were euthymic and taking antidepressants at the beginning of pregnancy. Consider the risk of untreated depression when discontinuing or changing treatment with antidepressant medication during pregnancy and postpartum.
Pregnant women with fibromyalgia are at increased risk for adverse maternal and infant outcomes including preterm premature rupture of membranes, preterm birth, small for gestational age, intrauterine growth restriction, placental disruption, and venous thrombosis. It is not known if these adverse maternal and fetal outcomes are a direct result of fibromyalgia or other comorbid factors.
Maternal Adverse Reactions
Use of CYMBALTA in the month before delivery may be associated with an increased risk of postpartum hemorrhage [see Warnings and Precautions (5.5)].
Fetal/Neonatal Adverse Reaction
Neonates exposed to CYMBALTA and other SNRIs or SSRIs late in the third trimester have developed complications requiring prolonged hospitalization, respiratory support, and tube feeding. Such complications can arise immediately upon delivery. Reported clinical findings have included respiratory distress, cyanosis, apnea, seizures, temperature instability, feeding difficulty, vomiting, hypoglycemia, hypotonia, hypertonia, hyperreflexia, tremor, jitteriness, irritability, and constant crying. These findings are consistent with either a direct toxic effect of the SNRIs or SSRIs, or possibly, a drug discontinuation syndrome. It should be noted that, in some cases, the clinical picture is consistent with serotonin syndrome [see Warnings and Precautions (5.4)].
Data
Human Data
Data from a postmarketing retrospective claims-based cohort study found an increased risk for postpartum hemorrhage among 955 pregnant women exposed to duloxetine in the last month of pregnancy compared to 4,128,460 unexposed pregnant women (adjusted relative risk: 1.53; 95% CI: 1.08-2.18). The same study did not find a clinically meaningful increase in the risk for major birth defects in the comparison of 2532 women exposed to duloxetine in the first trimester of pregnancy to 1,284,827 unexposed women after adjusting for several confounders. Methodologic limitations include possible residual confounding, misclassification of exposure and outcomes, lack of direct measures of disease severity, and lack of information about alcohol use, nutrition, and over-the-counter medication exposures.
Animal Data
In animal reproduction studies, duloxetine has been shown to have adverse effects on embryo/fetal and postnatal development.
When duloxetine was administered orally to pregnant rats and rabbits during the period of organogenesis, there was no evidence of malformations or developmental variations at doses up to 45 mg/kg/day [3 and 6 times, respectively, the MRHD of 120 mg/day given to adolescents on a mg/m2 basis]. However, fetal weights were decreased at this dose, with a no-effect dose of 10 mg/kg/day (approximately equal to the MRHD in rats and 2 times the MRHD in rabbits).
When duloxetine was administered orally to pregnant rats throughout gestation and lactation, the survival of pups to 1 day postpartum and pup body weights at birth and during the lactation period were decreased at a dose of 30 mg/kg/day (2 times the MRHD given to adolescents on a mg/m2 basis); the no-effect dose was 10 mg/kg/day. Furthermore, behaviors consistent with increased reactivity, such as increased startle response to noise and decreased habituation of locomotor activity, were observed in pups following maternal exposure to 30 mg/kg/day. Post-weaning growth and reproductive performance of the progeny were not affected adversely by maternal duloxetine treatment.
8.2 Lactation
Risk Summary
Data from published literature report the presence of duloxetine in human milk (see Data). There are reports of sedation, poor feeding, and poor weight gain in infants exposed to duloxetine through breast milk (see Clinical Considerations). There are no data on the effect of duloxetine on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for CYMBALTA and any potential adverse effects on the breastfed child from CYMBALTA or from the underlying maternal condition.
Clinical Considerations
Infants exposed to CYMBALTA should be monitored for sedation, poor feeding and poor weight gain.
Data
Disposition of CYMBALTA was studied in 6 lactating women who were at least 12 weeks postpartum and had elected to wean their infants. The women were given 40 mg of CYMBALTA twice daily for 3.5 days. The peak concentration measured in breast milk occurred at a median of 3 hours after the dose. The amount of CYMBALTA in breast milk was approximately 7 mcg/day while on that dose; the estimated daily infant dose was approximately 2 mcg/kg/day, which is less than 1% of the maternal dose. The presence of CYMBALTA metabolites in breast milk was not examined.
8.4 Pediatric Use
The safety and effectiveness of CYMBALTA have been established for treatment of generalized anxiety disorder (GAD) in patients 7 to 17 years of age and for treatment of juvenile fibromyalgia syndrome in patients 13 to 17 years of age. The safety and effectiveness of CYMBALTA have not been established in pediatric patients with major depressive disorder (MDD), diabetic peripheral neuropathic pain, or chronic musculoskeletal pain.
Antidepressants increased the risk of suicidal thoughts and behavior in pediatric patients. Monitor all pediatric patients being treated with antidepressants for clinical worsening and emergence of suicidal thoughts and behaviors, especially during the initial few months of treatment, or at times of dosage changes [see Warnings and Precautions (5.1)]. Perform regular monitoring of weight and growth in pediatric patients treated with CYMBALTA [see Adverse Reactions (6.1)].
Generalized Anxiety Disorder
Use of CYMBALTA for the treatment of GAD in patients 7 to 17 years of age is supported by one 10-week, placebo-controlled trial (GAD-6). The study included 272 pediatric patients with GAD of which 47% were 7 to 11 years of age (53% were 12 to 17 years of age). CYMBALTA demonstrated superiority over placebo as measured by greater improvement in the Pediatric Anxiety Rating Scale (PARS) for GAD severity score [see Clinical Studies (14.3)].
The safety and effectiveness of CYMBALTA for the treatment of GAD in pediatric patients less than 7 years of age have not been established.
Fibromyalgia
Use of CYMBALTA for treatment of fibromyalgia in patients 13 to 17 years of age is supported by a 13-week placebo-controlled trial in 184 patients with juvenile fibromyalgia syndrome (Study FM-4). CYMBALTA showed improvement over placebo on the primary endpoint, change from baseline to end-of-treatment on the Brief Pain Inventory (BPI) – Modified Short Form: Adolescent Version 24-hour average pain severity rating [see Clinical Studies (14.5)].
The safety and effectiveness of CYMBALTA for the treatment of fibromyalgia in patients less than 13 years of age have not been established.
Major Depressive Disorder
The safety and effectiveness of CYMBALTA have not been established in pediatric patients for the treatment of MDD. Efficacy of CYMBALTA was not demonstrated in two 10-week, placebo-controlled trials with 800 pediatric patients aged 7 to 17 years old with MDD (MDD-6 and MDD-7). Neither CYMBALTA nor an active control (approved for treatment of pediatric MDD) was superior to placebo.
The most frequently observed adverse reactions in the MDD pediatric clinical trials included nausea, headache, decreased weight, and abdominal pain. Decreased appetite and weight loss have been observed in association with the use of SSRIs and SNRIs.
Juvenile Animal Toxicology Data
Duloxetine administration to young rats from post-natal day 21 (weaning) through post-natal day 90 (adult) resulted in decreased body weights that persisted into adulthood, but recovered when drug treatment was discontinued; slightly delayed (~1.5 days) sexual maturation in females, without any effect on fertility; and a delay in learning a complex task in adulthood, which was not observed after drug treatment was discontinued. These effects were observed at the high dose of 45 mg/kg/day (2 times the MRHD, for a child); the no-effect-level was 20 mg/kg/day (≈1 times the MRHD, for a child).
8.5 Geriatric Use
Geriatric Exposure in Premarketing Clinical Trials of CYMBALTA
- Of the 2,418 patients in MDD trials, 6% (143) were 65 years of age or over.
- Of the 1041 patients in CLBP trials, 21% (221) were 65 years of age or over.
- Of the 487 patients in OA trials, 41% (197) were 65 years of age or over.
- Of the 1,074 patients in the DPNP trials, 33% (357) were 65 years of age or over.
- Of the 1,761 patients in FM trials, 8% (140) were 65 years of age or over.
In the MDD, GAD, DPNP, FM, OA, and CLBP studies, no overall differences in safety or effectiveness were generally observed between these patients and younger adult patients, and other reported clinical experience has not identified differences in responses between these geriatric and younger adult patients, but greater sensitivity of some older patients cannot be ruled out.
SSRIs and SNRIs, including CYMBALTA have been associated with clinically significant hyponatremia in geriatric patients, who may be at greater risk for this adverse reaction [see Warnings and Precautions (5.13)].
In an analysis of data from all placebo-controlled-trials, CYMBALTA-treated patients reported a higher rate of falls compared to placebo-treated patients. The increased risk appears to be proportional to a patient's underlying risk for falls. Underlying risk appears to increase steadily with age. As geriatric patients tend to have a higher prevalence of risk factors for falls such as medications, medical comorbidities and gait disturbances, the impact of increasing age by itself on falls during CYMBALTA treatment is unclear. Falls with serious consequences including bone fractures and hospitalizations have been reported with CYMBALTA use [see Warnings and Precautions (5.3) and Adverse Reactions (6.1)].
The pharmacokinetics of duloxetine after a single dose of 40 mg were compared in healthy elderly females (65 to 77 years) and healthy middle-age females (32 to 50 years). There was no difference in the Cmax, but the AUC of duloxetine was somewhat (about 25%) higher and the half-life about 4 hours longer in the elderly females. Population pharmacokinetic analyses suggest that the typical values for clearance decrease by approximately 1% for each year of age between 25 to 75 years of age; but age as a predictive factor only accounts for a small percentage of between-patient variability. Dosage adjustment based on the age of the adult patient is not necessary.
8.6 Gender
Duloxetine's half-life is similar in men and women. Dosage adjustment based on gender is not necessary.
8.7 Smoking Status
Duloxetine bioavailability (AUC) appears to be reduced by about one-third in smokers. Dosage modifications are not recommended for smokers.
8.9 Hepatic Impairment
Patients with clinically evident hepatic impairment have decreased duloxetine metabolism and elimination. After a single 20 mg dose of CYMBALTA, 6 cirrhotic patients with moderate liver impairment (Child-Pugh Class B) had a mean plasma duloxetine clearance about 15% that of age- and gender-matched healthy subjects, with a 5-fold increase in mean exposure (AUC). Although Cmax was similar to normals in the cirrhotic patients, the half-life was about 3 times longer [see Dosage and Administration (2.7) and Warnings and Precautions (5.14)].
8.10 Severe Renal Impairment
Limited data are available on the effects of CYMBALTA in patients with end-stage renal disease (ESRD). After a single 60 mg dose of CYMBALTA, Cmax and AUC values were approximately 100% greater in patients with ESRD receiving chronic intermittent hemodialysis than in subjects with normal renal function. The elimination half-life, however, was similar in both groups. The AUCs of the major circulating metabolites, 4-hydroxy duloxetine glucuronide and 5-hydroxy, 6-methoxy duloxetine sulfate, largely excreted in urine, were approximately 7- to 9-fold higher and would be expected to increase further with multiple dosing. Population PK analyses suggest that mild to moderate degrees of renal impairment (estimated CrCl 30-80 mL/min) have no significant effect on duloxetine apparent clearance [see Dosage and Administration (2.7) and Warnings and Precautions (5.14)].
9 DRUG ABUSE AND DEPENDENCE
9.2 Abuse
In animal studies, duloxetine did not demonstrate barbiturate-like (depressant) abuse potential.
While CYMBALTA has not been systematically studied in humans for its potential for abuse, there was no indication of drug-seeking behavior in the clinical trials. However, it is not possible to predict on the basis of premarketing experience the extent to which a CNS active drug will be misused, diverted, and/or abused once marketed. Consequently, physicians should carefully evaluate patients for a history of drug abuse and follow such patients closely, observing them for signs of misuse or abuse of CYMBALTA (e.g., development of tolerance, incrementation of dose, drug-seeking behavior).
10 OVERDOSAGE
10.1 Signs and Symptoms
In postmarketing experience, fatal outcomes have been reported for acute CYMBALTA overdoses, primarily with mixed overdoses, but also with CYMBALTA only, including 1000 mg of CYMBALTA (approximately 8.3 times the maximum recommended dosage). Signs and symptoms of overdose (CYMBALTA alone or with mixed drugs) included somnolence, coma, serotonin syndrome, seizures, syncope, tachycardia, hypotension, hypertension, and vomiting.
10.2 Management of Overdose
There is no specific antidote to a CYMBALTA overdosage, but if serotonin syndrome ensues, specific treatment (such as with cyproheptadine and/or temperature control) may be considered.
In case of acute overdose with CYMBALTA, treatment should consist of those general measures employed in the management of overdose with any drug, such as assuring an adequate airway, oxygenation, and ventilation and monitoring cardiac rhythm and vital signs. Gastric lavage with a large-bore orogastric tube with appropriate airway protection, if needed, may be indicated if performed soon after ingestion or in symptomatic patients. Induction of emesis is not recommended.
Activated charcoal may be useful in limiting absorption of duloxetine from the gastrointestinal tract. Administration of activated charcoal has been shown to decrease duloxetine AUC and Cmax by an average of one-third, although some patients had a limited effect of activated charcoal. Due to the large volume of distribution of duloxetine, forced diuresis, dialysis, hemoperfusion, and exchange transfusion are unlikely to be beneficial.
In managing overdose, the possibility of multiple drug involvement should be considered. A specific caution involves patients who overdose with CYMBALTA and tricyclic antidepressants. In such a case, decreased clearance of the parent tricyclic and/or its active metabolite may increase the possibility of clinically significant sequelae and extend the time needed for close medical observation [see Warnings and Precautions (5.4) and Drug Interactions (7)].
Consider contacting a poison control center (1-800-222-1222 or www.poison.org) for additional information on the treatment of overdosage.
11 DESCRIPTION
CYMBALTA® (duloxetine delayed-release capsules) is a selective serotonin and norepinephrine reuptake inhibitor (SNRI) for oral administration. Its chemical designation is (+)-(S)-N-methyl-γ-(1-naphthyloxy)-2-thiophenepropylamine hydrochloride. The empirical formula is C18H19NOS•HCl, which corresponds to a molecular weight of 333.88. The structural formula is:
Duloxetine hydrochloride is a white to slightly brownish white solid, which is slightly soluble in water.
Each capsule contains enteric-coated pellets of 20, 30, or 60 mg of duloxetine (equivalent to 22.4, 33.7, or 67.3 mg of duloxetine hydrochloride, respectively). These enteric-coated pellets are designed to prevent degradation of the drug in the acidic environment of the stomach. Inactive ingredients include FD&C Blue No. 2, gelatin, hypromellose, hydroxypropyl methylcellulose acetate succinate, sodium lauryl sulfate, sucrose, sugar spheres, talc, titanium dioxide, and triethyl citrate. The 20 and 60 mg capsules also contain iron oxide yellow.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Although the exact mechanisms of the antidepressant, central pain inhibitory and anxiolytic actions of duloxetine in humans are unknown, these actions are believed to be related to its potentiation of serotonergic and noradrenergic activity in the CNS.
12.2 Pharmacodynamics
Preclinical studies have shown that duloxetine is a potent inhibitor of neuronal serotonin and norepinephrine reuptake and a less potent inhibitor of dopamine reuptake. Duloxetine has no significant affinity for dopaminergic, adrenergic, cholinergic, histaminergic, opioid, glutamate, and GABA receptors in vitro. Duloxetine does not inhibit monoamine oxidase (MAO).
CYMBALTA is in a class of drugs known to affect urethral resistance [see Warnings and Precautions (5.15)].
Cardiac Electrophysiology
The effect of CYMBALTA 160 mg and 200 mg administered twice daily (2.7 and 3.3 times the maximum recommended dosage, respectively) to steady state was evaluated in a randomized, double-blinded, two-way crossover study in 117 healthy female adult subjects. No QT interval prolongation was detected. CYMBALTA appears to be associated with concentration-dependent but not clinically meaningful QT shortening.
12.3 Pharmacokinetics
Duloxetine has an elimination half-life of about 12 hours (range 8 to 17 hours) and its pharmacokinetics are dose proportional over the therapeutic range. Steady-state plasma concentrations are typically achieved after 3 days of dosing. Elimination of duloxetine is mainly through hepatic metabolism involving two P450 isozymes, CYP1A2 and CYP2D6.
Absorption
After oral CYMBALTA administration, duloxetine hydrochloride is well absorbed. There is a median 2 hour lag until absorption begins (Tlag), with maximal plasma concentrations (Cmax) of duloxetine occurring 6 hours post dose. There is a 3 hour delay in absorption and a one-third increase in apparent clearance of duloxetine after an evening dose as compared to a morning dose.
Distribution
The apparent volume of distribution averages about 1640 L. Duloxetine is highly bound (>90%) to proteins in human plasma, binding primarily to albumin and α1-acid glycoprotein. The interaction between duloxetine and other highly protein bound drugs has not been fully evaluated. Plasma protein binding of duloxetine is not affected by renal or hepatic impairment.
Elimination
Metabolism
Biotransformation and disposition of duloxetine in humans have been determined following oral administration of 14C-labeled duloxetine. Duloxetine comprises about 3% of the total radiolabeled material in the plasma, indicating that it undergoes extensive metabolism to numerous metabolites. The major biotransformation pathways for duloxetine involve oxidation of the naphthyl ring followed by conjugation and further oxidation. Both CYP1A2 and CYP2D6 catalyze the oxidation of the naphthyl ring in vitro. Metabolites found in plasma include 4-hydroxy duloxetine glucuronide and 5-hydroxy, 6-methoxy duloxetine sulfate.
Excretion
Many additional metabolites have been identified in urine, some representing only minor pathways of elimination. Only trace (<1% of the dose) amounts of unchanged duloxetine are present in the urine. Most (about 70%) of the duloxetine dose appears in the urine as metabolites of duloxetine; about 20% is excreted in the feces. Duloxetine undergoes extensive metabolism, but the major circulating metabolites have not been shown to contribute significantly to the pharmacologic activity of duloxetine.
Specific Populations
Pediatric Patients
Duloxetine steady-state plasma concentration was comparable in pediatric patients 7 to 17 years of age and adult patients. The average steady-state duloxetine concentration was approximately 30% lower in this pediatric population relative to adult patients. The model-predicted duloxetine steady state plasma concentrations in pediatric patients 7 to 17 years of age were mostly within the concentration range observed in adult patients and did not exceed the concentration range in adults.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Duloxetine was administered in the diet to mice and rats for 2 years.
In female mice receiving duloxetine at 140 mg/kg/day (3 times the maximum recommended human dose (MRHD) of 120 mg/day given to children on a mg/m2 basis), there was an increased incidence of hepatocellular adenomas and carcinomas. The no-effect dose was 50 mg/kg/day (1 time the MRHD given to children). Tumor incidence was not increased in male mice receiving duloxetine at doses up to 100 mg/kg/day (2 times the MRHD given to children).
In rats, dietary doses of duloxetine up to 27 mg/kg/day in females (1 time the MRHD given to children) and up to 36 mg/kg/day in males (1.4 times the MRHD given to children) did not increase the incidence of tumors.
Mutagenesis
Duloxetine was not mutagenic in the in vitro bacterial reverse mutation assay (Ames test) and was not clastogenic in an in vivo chromosomal aberration test in mouse bone marrow cells. Additionally, duloxetine was not genotoxic in an in vitro mammalian forward gene mutation assay in mouse lymphoma cells or in an in vitro unscheduled DNA synthesis (UDS) assay in primary rat hepatocytes, and did not induce sister chromatid exchange in Chinese hamster bone marrow in vivo.
14 CLINICAL STUDIES
14.1 Overview of the Clinical Trials
The efficacy of CYMBALTA has been established in the following populations in adequate and well-controlled trials:
- Major Depressive Disorder (MDD): 4 short-term (Studies MDD-1, MDD-2, MDD-3, and MDD-4) and 1 maintenance trial (Study MDD-5) in adults [see Clinical Studies (14.2)].
- Generalized Anxiety Disorder (GAD): 3 short-term trials in adults (Studies GAD-1, GAD-2, and GAD-3), 1 maintenance trial in adults (Study GAD-4), 1 short-term trial in geriatric patients (Study GAD-5), and 1 short-term trial in pediatric patients 7 to 17 years of age (Study GAD-6) [see Clinical Studies (14.3)].
- Diabetic Peripheral Neuropathic Pain (DPNP): Two 12-week trials in adults (Studies DPNP-1 and DPNP-2) [see Clinical Studies (14.4)].
- Fibromyalgia (FM): Two trials in adults (one of 3 months duration and one of 6 months duration) (Studies FM-1 and FM-2) and one 13-week trial in pediatric patients 13 to 17 years of age (Study FM-4) [see Clinical Studies (14.5)].
- Chronic Musculoskeletal Pain: Two 12- to 13-week trials in adult patients with chronic low back pain (CLBP) (Studies CLBP-1 and CLBP-3) and one 13-week trial in adult patients with chronic pain due to osteoarthritis (OA) (Study OA-1) [see Clinical Studies (14.6)].
Additionally, a summary of the following trials that did not demonstrate efficacy are presented below: Study FM-3 (a 16- week trial in adult patients with fibromyalgia), Study CLBP-2 (a 13-week trial in adult patients with CLBP), and Study OA-2 (a 13-week trial in adult patients with chronic pain due to OA).
14.2 Major Depressive Disorder in Adults
The efficacy of CYMBALTA as a treatment for MDD in adults was established in 4 randomized, double-blind, placebo-controlled, fixed-dose trials in adult outpatients (18 to 83 years) meeting DSM-IV criteria for MDD:
- In Studies MDD-1 and MDD-2, patients were randomized to CYMBALTA 60 mg once daily (N=123 and N=128, respectively) or placebo (N=122 and N=139, respectively) for 9 weeks
- In Study MDD-3, patients were randomized to CYMBALTA 20 or 40 mg twice daily (N=86 and N=91, respectively) or placebo (N=89) for 8 weeks
- In Study MDD-4, patients were randomized to CYMBALTA 40 or 60 mg twice daily (N=95 and N=93, respectively) or placebo (N=93) for 8 weeks.
In all four trials, CYMBALTA demonstrated superiority over placebo as measured by improvement in the 17-item Hamilton Depression Rating Scale (HAMD-17) total score (see Table 8). There is no evidence that doses greater than 60 mg/day confer additional benefits.
In all of these clinical trials, analyses of the relationship between treatment outcome and age, gender, and race did not suggest any differential responsiveness on the basis of these patient characteristics.
|
SD: standard deviation; SE: standard error; LS Mean: least-squares mean; CI: confidence interval, not adjusted for multiplicity in trials where multiple dose groups were included. |
||||
|
a Difference (drug minus placebo) in least-squares mean change from baseline. |
||||
|
b Doses statistically significantly superior to placebo. |
||||
| Study Number | Treatment Group | Primary Efficacy Measure: HAMD-17 | ||
| Mean Baseline Score (SD) | LS Mean Change from Baseline (SE) | Placebo-subtracted Differencea (95% CI) | ||
| Study MDD-1 | CYMBALTA (60 mg/day)b | 21.5 (4.10) | -10.9 (0.70) | -4.9 (-6.8, -2.9) |
| Placebo | 21.1 (3.71) | -6.1 (0.69) | -- | |
| Study MDD-2 | CYMBALTA (60 mg/day)b | 20.3 (3.32) | -10.5 (0.71) | -2.2 (-4.0, -0.3) |
| Placebo | 20.5 (3.42) | -8.3 (0.67) | -- | |
| Study MDD-3 | CYMBALTA (20 mg BID)b | 18.6 (5.85) | -7.4 (0.80) | -2.4 (-4.7, -0.2) |
| CYMBALTA (40 mg BID)b | 18.1 (4.52) | -8.6 (0.81) | -3.6 (-5.9, -1.4) | |
| Placebo | 17.2 (5.11) | -5.0 (0.81) | -- | |
| Study MDD-4 | CYMBALTA (40 mg BID)b | 19.9 (3.54) | -11.0 (0.49) | -2.2 (-3.6, -0.9) |
| CYMBALTA (60 mg BID)b | 20.2 (3.41) | -12.1 (0.49) | -3.3 (-4.7, -1.9) | |
| Placebo | 19.9 (3.58) | -8.8 (0.50) | -- | |
In Study MDD-5, 533 adult patients meeting DSM-IV criteria for MDD received CYMBALTA 60 mg once daily during an initial 12-week open-label treatment phase. Two hundred and seventy-eight patients who responded to open label treatment [defined as meeting the following criteria at weeks 10 and 12: a HAMD-17 total score ≤9, Clinical Global Impressions of Severity (CGI-S) ≤2, and not meeting the DSM-IV criteria for MDD] were randomly assigned to continuation of CYMBALTA at the same dosage (N=136) or to placebo (N=142) for 6 months.
In Study MDD-5, patients on CYMBALTA experienced a statistically significantly longer time to relapse of depression than did patients on placebo (see Figure 1). Relapse was defined as an increase in the CGI-S score of ≥2 points compared with that obtained at week 12, as well as meeting the DSM-IV criteria for MDD at 2 consecutive visits at least 2 weeks apart, where the 2-week temporal criterion had to be satisfied at only the second visit.
Figure 1: Cumulative Proportiona of Adult Patients with MDD Relapse (Study MDD-5)
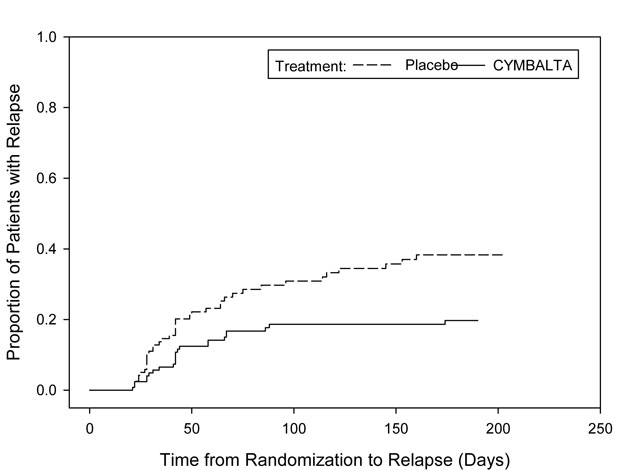
a Kaplan-Meier estimator method.
14.3 Generalized Anxiety Disorder
GAD Trials in Adults (Including Geriatric Patients)
The efficacy of CYMBALTA in the treatment of generalized anxiety disorder (GAD) was established in 1 fixed-dose randomized, double-blind, placebo-controlled trial and 2 flexible-dose randomized, double-blind, placebo-controlled trials in adult outpatients between 18 and 83 years of age meeting the DSM-IV criteria for GAD (Studies GAD-1, GAD-2, and GAD-3, respectively).
In Studies GAD-1 and GAD-2, the starting dose was 60 mg once daily (down titration to 30 mg once daily was allowed for tolerability reasons; the dosage could be increased to 60 mg once daily). Fifteen percent of patients were down titrated. Study GAD-3 had a starting dose of 30 mg once daily for 1 week before increasing it to 60 mg once daily.
Studies GAD-2 and GAD-3 involved dose titration with CYMBALTA doses ranging from 60 mg once daily to 120 mg once daily (N=168 and N=162) compared to placebo (N=159 and N=161) over a 10-week treatment period. The mean dosage for completers at endpoint in these trials was 104.8 mg/day. Study GAD-1 evaluated CYMBALTA dosages of 60 mg once daily (N=168) and 120 mg once daily (N=170) compared to placebo (N=175) over a 9-week treatment period. While a 120 mg/day dose was shown to be effective, there is no evidence that doses greater than 60 mg/day confer additional benefit.
In all 3 trials, CYMBALTA demonstrated superiority over placebo as measured by greater improvement in the Hamilton Anxiety Scale (HAM-A) total score (see Table 8) and by the Sheehan Disability Scale (SDS) global functional impairment score. The SDS is a composite measurement of the extent emotional symptoms disrupt patient functioning in 3 life domains: work/school, social life/leisure activities, and family life/home responsibilities.
In Study GAD-4, 887 patients meeting DSM-IV-TR criteria for GAD received CYMBALTA 60 mg to 120 mg once daily during an initial 26-week open-label treatment phase. Four hundred and twenty-nine patients who responded to open-label treatment [defined as meeting the following criteria at weeks 24 and 26: a decrease from baseline HAM-A total score by at least 50% to a score no higher than 11, and a Clinical Global Impressions of Improvement (CGI-Improvement) score of 1 or 2] were randomly assigned to continuation of CYMBALTA at the same dosage (N=216) or to placebo (N=213) and were observed for relapse. Of the patients randomized, 73% had been in a responder status for at least 10 weeks. Relapse was defined as an increase in CGI-Severity score at least 2 points to a score ≥4 and a MINI (Mini-International Neuropsychiatric Interview) diagnosis of GAD (excluding duration), or discontinuation due to lack of efficacy. Patients taking CYMBALTA experienced a statistically significantly longer time to relapse of GAD than did patients taking placebo (see Figure 2).
Subgroup analyses did not indicate that there were any differences in treatment outcomes as a function of age or gender.
GAD Trial in Geriatric Patients
The efficacy of CYMBALTA in the treatment of patients ≥65 years of age with GAD was established in one 10-week flexible-dose, randomized, double-blind, placebo-controlled trial in adults ≥65 years of age meeting the DSM-IV criteria for GAD (Study GAD-5). In Study GAD-5, the starting dose was 30 mg once daily for 2 weeks before further dose increases in 30 mg increments at treatment weeks 2, 4, and 7 up to 120 mg once daily were allowed based on investigator judgment of clinical response and tolerability. The mean dosage for patients completing the 10-week acute treatment phase was 51 mg. Patients treated with CYMBALTA (N=151) demonstrated significantly greater improvement compared with placebo (N=140) on mean change from baseline to endpoint as measured by the HAM-A total score (see Table 8).
GAD Trial in Pediatric Patients 7 to 17 Years Old
The efficacy of CYMBALTA in the treatment of pediatric patients 7 to 17 years of age with GAD was established in 1 flexible-dose randomized, double-blind, placebo-controlled trial in pediatric outpatients with GAD (based on DSM-IV criteria) (Study GAD-6).
In Study GAD-6, the starting dosage was 30 mg once daily for 2 weeks. Further dosage increases in 30 mg increments up to 120 mg once daily were allowed based on investigator judgment of clinical response and tolerability. The mean dosage for patients completing the 10-week treatment phase was 57.6 mg/day. In this study, CYMBALTA (N=135) demonstrated superiority over placebo (N=137) from baseline to endpoint as measured by greater improvement in the Pediatric Anxiety Rating Scale (PARS) for GAD severity score (see Table 9).
|
SD: standard deviation; SE: standard error; LS Mean: least-squares mean; CI: confidence interval, not adjusted for multiplicity in trials where multiple dose groups were included. |
||||
|
a Difference (drug minus placebo) in least squares mean change from baseline. |
||||
|
b Dose statistically significantly superior to placebo. |
||||
| Study Number (population) (measurement) |
Treatment Group | Primary Efficacy Measure | ||
| Mean Baseline Score (SD) | LS Mean Change from Baseline (SE) | Placebo-subtracted Differencea (95% CI) | ||
| Study GAD-1 (Adult) (HAM-A) |
CYMBALTA (60 mg/day)b | 25.1 (7.18) | -12.8 (0.68) | -4.4 (-6.2, -2.5) |
| CYMBALTA (120 mg/day)b | 25.1 (7.24) | -12.5 (0.67) | -4.1 (-5.9, -2.3) | |
| Placebo | 25.8 (7.66) | -8.4 (0.67) | -- | |
| Study GAD-2 (Adult) (HAM-A) |
CYMBALTA (60-120 mg/day)b | 22.5 (7.44) | -8.1 (0.70) | -2.2 (-4.2, -0.3) |
| Placebo | 23.5 (7.91) | -5.9 (0.70) | -- | |
| Study GAD-3 (Adult) (HAM-A) |
CYMBALTA (60-120 mg/day)b | 25.8 (5.66) | -11.8 (0.69) | -2.6 (-4.5, -0.7) |
| Placebo | 25.0 (5.82) | -9.2 (0.67) | -- | |
| Study GAD-5 (Geriatric) (HAM-A) |
CYMBALTA (60-120 mg/day)b | 24.6 (6.21) | -15.9 (0.63) | -4.2 (-5.9, -2.5) |
| Placebo | 24.5 (7.05) | -11.7 (0.67) | -- | |
| Study GAD-6 (Pediatric) (PARS for GAD) |
CYMBALTA (30-120 mg/day)b | 17.5 (1.98) | -9.7 (0.50) | -2.7 (-4.0, -1.3) |
| Placebo | 17.4 (2.24) | -7.1 (0.50) | -- | |
Figure 2: Cumulative Proportiona of Adult Patients with GAD Relapse (Study GAD-4)
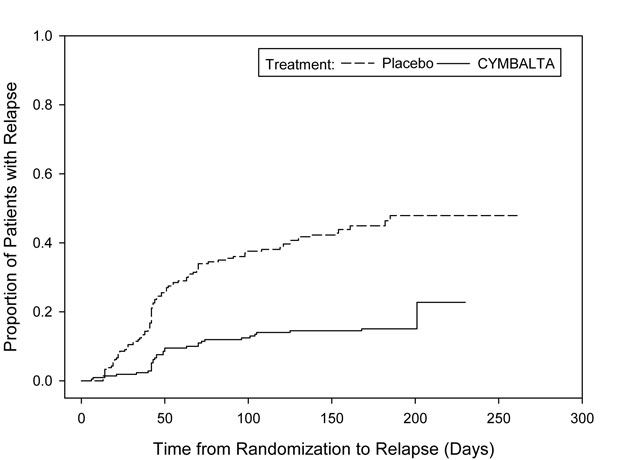
a Kaplan-Meier estimator method.
14.4 Diabetic Peripheral Neuropathic Pain in Adults
The efficacy of CYMBALTA for the management of neuropathic pain associated with diabetic peripheral neuropathy in adults was established in 2 randomized, 12-week, double-blind, placebo-controlled, fixed-dose trials in adult patients having diabetic peripheral neuropathic pain (DPNP) for at least 6 months (Study DPNP-1 and Study DPNP-2). These trials enrolled a total of 791 patients of whom 592 (75%) completed the trials. Patients enrolled had Type I or II diabetes mellitus with a diagnosis of painful distal symmetrical sensorimotor polyneuropathy for at least 6 months. The patients had a baseline pain score of ≥4 on an 11-point scale ranging from 0 (no pain) to 10 (worst possible pain). Patients were permitted up to 4 grams of acetaminophen per day as needed for pain, in addition to CYMBALTA. Patients recorded their pain daily in a diary.
Both trials compared CYMBALTA 60 mg once daily or 60 mg twice daily with placebo. Study DPNP-1 additionally compared CYMBALTA 20 mg with placebo. A total of 457 patients (342 CYMBALTA, 115 placebo) were enrolled in Study DPNP-1 and a total of 334 patients (226 CYMBALTA, 108 placebo) were enrolled in Study DPNP-2.
Treatment with CYMBALTA 60 mg one or two times a day statistically significantly improved the endpoint mean pain scores from baseline and increased the proportion of patients with at least a 50% reduction in pain scores from baseline. For various degrees of improvement in pain from baseline to study endpoint, Figures 3 and 4 show the fraction of patients achieving that degree of improvement in Studies DPNP-1 and DPNP-2, respectively. The figures are cumulative, so that patients whose change from baseline is, for example, 50%, are also included at every level of improvement below 50%. Patients who did not complete the trial were assigned 0% improvement. Some patients experienced a decrease in pain as early as week 1, which persisted throughout the trial.
Figure 3: Percentage of DPNP Adult Patients Achieving Various Levels of Pain Relief as Measured by 24-Hour Average Pain Severity (Study DPNP-1)
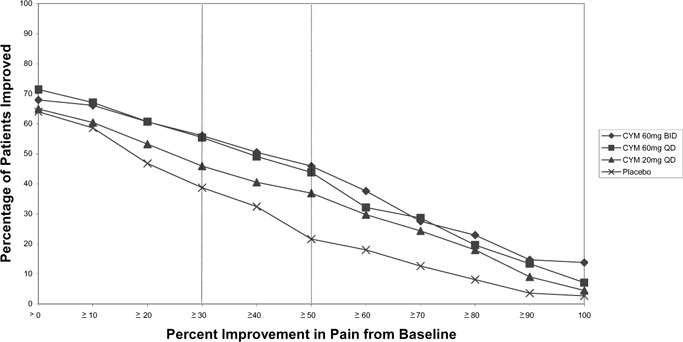
Figure 4: Percentage of DPNP Adult Patients Achieving Various Levels of Pain Relief as Measured by 24-Hour Average Pain Severity (Study DPNP-2)
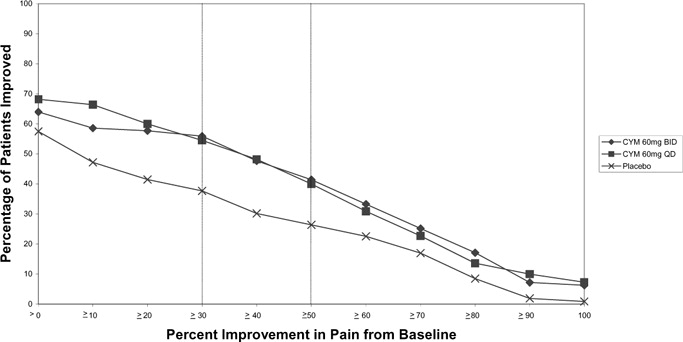
14.5 Fibromyalgia
Adult Trials in Fibromyalgia
The efficacy of CYMBALTA for the management of fibromyalgia in adults was established in two randomized, double-blind, placebo-controlled, fixed-dose trials in adult patients meeting the American College of Rheumatology criteria for fibromyalgia (a history of widespread pain for 3 months, and pain present at 11 or more of the 18 specific tender point sites). Study FM-1 was three months in duration and enrolled female patients only. Study FM-2 was six months in duration and enrolled male and female patients. Approximately 25% of participants had a comorbid diagnosis of MDD. Studies FM-1 and FM-2 enrolled a total of 874 patients of whom 541 (62%) completed the trials. A total of 354 patients (234 CYMBALTA, 120 placebo) were enrolled in Study FM-1 and a total of 520 patients (376 CYMBALTA, 144 placebo) were enrolled in Study FM-2 (5% male, 95% female). The patients had a baseline pain score of 6.5 on an 11-point scale ranging from 0 (no pain) to 10 (worse possible pain).
Studies FM-1 and FM-2 compared CYMBALTA 60 mg once daily or 120 mg daily (given in divided doses in Study FM-1 and as a single daily dose in Study FM-2) with placebo. Study FM-2 additionally compared CYMBALTA 20 mg with placebo during the initial three months of a six-month trial.
Treatment with CYMBALTA 60 mg or 120 mg daily statistically significantly improved the endpoint mean pain scores from baseline and increased the proportion of patients with at least a 50% reduction in pain score from baseline. Pain reduction was observed in patients both with and without comorbid MDD. However, the degree of pain reduction may be greater in patients with comorbid MDD. For various degrees of improvement in pain from baseline to study endpoint, Figures 5 and 6 show the fraction of patients achieving that degree of improvement in Studies FM-1 and FM-2, respectively. The figures are cumulative so that patients whose change from baseline is, for example, 50%, are also included at every level of improvement below 50%. Patients who did not complete the trial were assigned 0% improvement. Some patients experienced a decrease in pain as early as week 1, which persisted throughout the trial. Improvement was also demonstrated on measures of function (Fibromyalgia Impact Questionnaires) and patient global impression of change (PGI). Neither trial demonstrated a benefit of 120 mg compared to 60 mg, and a higher dosage was associated with more adverse reactions and premature discontinuations of treatment.
Figure 5: Percentage of Adult Fibromyalgia Patients Achieving Various Levels of Pain Relief at Study Endpoint as Measured by 24-Hour Average Pain Severity (Study FM-1)
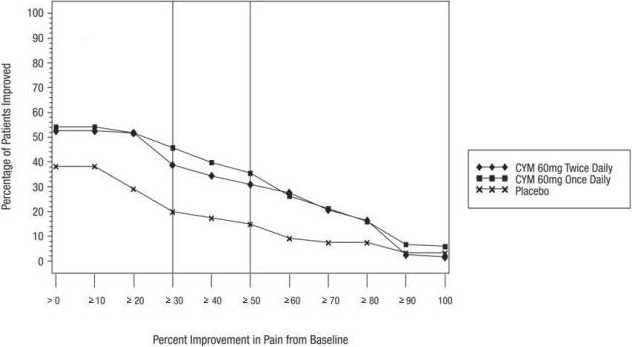
Figure 6: Percentage of Adult Fibromyalgia Patients Achieving Various Levels of Pain Relief at Study Endpoint as Measured by 24-Hour Average Pain Severity (Study FM-2)
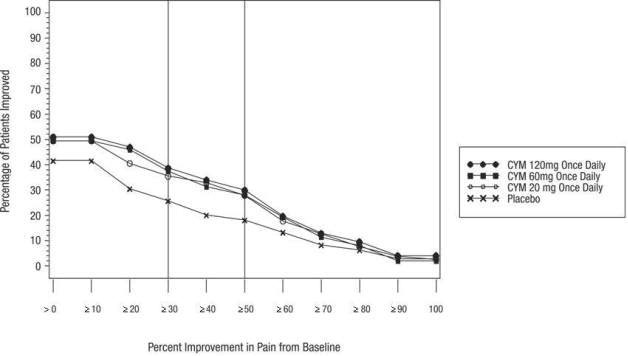
Additionally, the benefit of up-titration in non-responders to CYMBALTA at 60 mg/day was evaluated in a separate trial (Study FM-3). Adult patients were initially treated with CYMBALTA 60 mg once daily for eight weeks in open-label fashion. Subsequently, completers of this phase were randomized to double-blind treatment with CYMBALTA at either 60 mg once daily or 120 mg once daily. Responders were defined as patients who had at least a 30% reduction in pain score from baseline at the end of the 8-week treatment. Patients who were non-responders at 8 weeks were no more likely to meet response criteria at the end of 60 weeks of treatment if blindly titrated to CYMBALTA 120 mg as compared to those who were blindly continued on CYMBALTA 60 mg.
Pediatric Trial in Fibromyalgia
CYMBALTA was studied in 184 pediatric patients aged 13 to 17 years with juvenile fibromyalgia syndrome in a 13-week, placebo-controlled trial (Study FM-4). In Study FM-4, 149 (81%) patients completed the trial. CYMBALTA (N=91) was initiated at a dosage of 30 mg once daily for one week and titrated to 60 mg once daily for 12 weeks, as tolerated. The mean dosage for patients completing the 12-week treatment phase was 49 mg/day. CYMBALTA showed improvement over placebo on the primary endpoint [change from baseline to end-of-treatment on the Brief Pain Inventory (BPI) – Modified Short Form: Adolescent Version 24-hour average pain severity rating] with a p-value of 0.052 from the pre-specified primary analysis, and p-values ranging from 0.011-0.020 from sensitivity analyses which assigned baseline values to missing assessments of some patients who did not complete the trial for various reasons. The patients had a baseline BPI of 5.7. For various degrees of improvement in pain from baseline to study endpoint, Figure 7 shows the fraction of patients achieving that degree of improvement in Study FM-4.
Figure 7: Percentage of Pediatric Patients Aged 13 to 17 Years Old with Juvenile Fibromyalgia Syndrome Achieving Various Levels of Pain Relief at Week 12 (Study FM-4)a
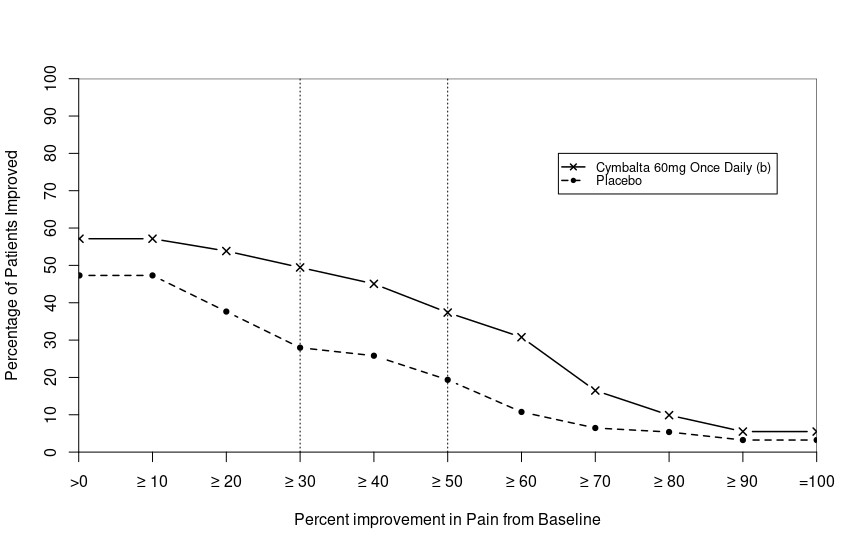
a Pain relief Measured by Brief Pain Inventory – Modified Short Form: Adolescent Version Average Pain Score.
b CYMBALTA-treated patients received 30 mg once daily for 1 week and subsequently titrated to 60 mg once daily for 12 weeks, as tolerated.
14.6 Chronic Musculoskeletal Pain in Adults
CYMBALTA is indicated for the treatment of chronic musculoskeletal pain in adults. This has been established in trials in adult patients with chronic low back pain and chronic pain due to osteoarthritis.
Trials in Chronic Low Back Pain in Adults
The efficacy of CYMBALTA in chronic low back pain (CLBP) in adults was assessed in two double-blind, placebo-controlled, randomized clinical trials of 13-weeks duration (Studies CLBP-1 and CLBP-2), and one of 12-weeks duration (CLBP-3). Studies CLBP-1 and CLBP-3 demonstrated efficacy of CYMBALTA in the treatment of CLBP. Patients in all trials had no signs of radiculopathy or spinal stenosis.
Study CLBP-1: Two hundred thirty-six adult patients (N=115 on CYMBALTA, N=121 on placebo) enrolled and 182 (77%) completed 13-week treatment phase. After 7 weeks of treatment, CYMBALTA-treated patients with less than 30% reduction in average daily pain and who were able to tolerate 60 mg once daily had their CYMBALTA dosage, in a double-blinded fashion, increased to 120 mg once daily for the remainder of the trial. Patients had a mean baseline pain rating of 6 on a numerical rating scale ranging from 0 (no pain) to 10 (worst possible pain). After 13 weeks of treatment, patients taking CYMBALTA 60-120 mg daily had a significantly greater pain reduction compared to patients taking placebo. Randomization was stratified by the patients' baseline NSAIDs use status. Subgroup analyses did not indicate that there were differences in treatment outcomes as a function of NSAIDs use.
Study CLBP-2: Four hundred and four patients were randomized to receive fixed dosages of CYMBALTA daily or a matching placebo (N=59 on CYMBALTA 20 mg, N=116 on CYMBALTA 60 mg, N=112 on CYMBALTA 120 mg, N=117 on placebo) and 267 (66%) completed the entire 13-week trial. After 13 weeks of treatment, none of the three CYMBALTA dosages showed a statistically significant difference in pain reduction compared to placebo.
Study CLBP-3: Four hundred and one patients were randomized to receive fixed doses of CYMBALTA 60 mg daily or placebo (N=198 on CYMBALTA, N=203 on placebo), and 303 (76%) completed the trial. Patients had a mean baseline pain rating of 6 on a numerical rating scale ranging from 0 (no pain) to 10 (worst possible pain). After 12 weeks of treatment, patients taking CYMBALTA 60 mg daily had significantly greater pain reduction compared to patients taking placebo.
For various degrees of improvement in pain from baseline to study endpoint, Figures 8 and 9 show the fraction of patients in Studies CLBP-1 and CLBP-3 achieving that degree of improvement, respectively. The figures are cumulative, so that patients whose change from baseline is, for example, 50%, are also included at every level of improvement below 50%. Patients who did not complete the trial were assigned the value of 0% improvement.
Figure 8: Percentage of Adult Patients with CLBP Achieving Various Levels of Pain Relief as Measured by 24-Hour Average Pain Severity (Study CLBP-1)
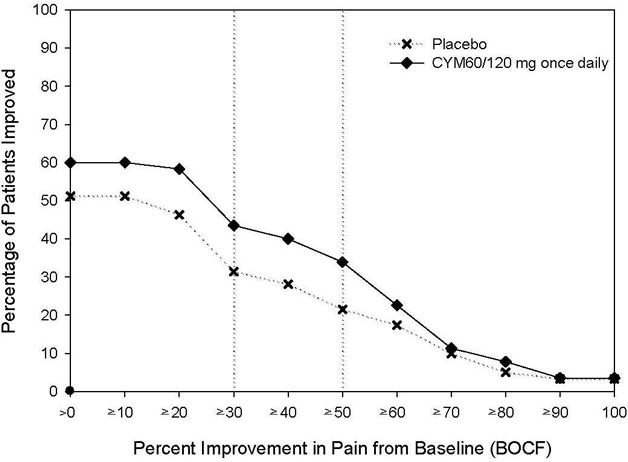
Figure 9: Percentage of Adult Patients with CLBP Achieving Various Levels of Pain Relief as Measured by 24-Hour Average Pain Severity (Study CLBP-3)
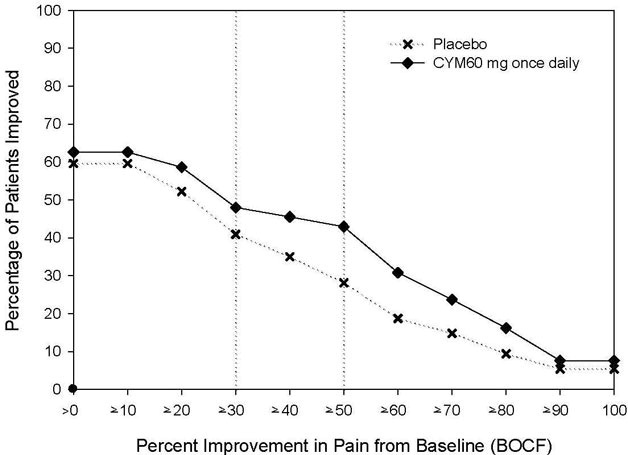
Trials in Chronic Pain Due to Osteoarthritis in Adults
The efficacy of CYMBALTA in chronic pain due to osteoarthritis (OA) in adults was assessed in 2 double-blind, placebo-controlled, randomized clinical trials of 13-weeks duration (Study OA-1 and Study OA-2). All patients in both trials fulfilled the ACR clinical and radiographic criteria for classification of idiopathic OA of the knee. Randomization was stratified by the patients' baseline NSAIDs-use status.
Patients assigned to CYMBALTA started treatment in both trials at a dose of 30 mg once daily for one week. After the first week, the dose of CYMBALTA was increased to 60 mg once daily. After 7 weeks of treatment with CYMBALTA 60 mg once daily, in Study OA-1 patients with sub-optimal response to treatment (<30% pain reduction) and tolerated CYMBALTA 60 mg once daily had their dose increased to 120 mg. However, in Study OA-2, all patients, regardless of their response to treatment after 7 weeks, were re-randomized to either continue receiving CYMBALTA 60 mg once daily or have their dosage increased to 120 mg once daily for the remainder of the trial. Patients in the placebo treatment groups in both trials received a matching placebo for the entire duration of trials. For both trials, efficacy analyses were conducted using 13-week data from the combined CYMBALTA 60 mg and 120 mg once daily treatment groups compared to the placebo group.
Study OA-1: Two hundred fifty-six patients (N=128 on CYMBALTA, N=128 on placebo) enrolled and 204 (80%) completed the trial. Patients had a mean baseline pain rating of 6 on a numerical rating scale ranging from 0 (no pain) to 10 (worst possible pain). After 13 weeks of treatment, patients taking CYMBALTA had significantly greater pain reduction than patients taking placebo. Subgroup analyses did not indicate that there were differences in treatment outcomes as a function of NSAIDs use.
Study OA-2: Two hundred thirty-one patients (N=111 on CYMBALTA, N=120 on placebo) enrolled and 173 (75%) completed the trial. Patients had a mean baseline pain of 6 on a numerical rating scale ranging from 0 (no pain) to 10 (worst possible pain). After 13 weeks of treatment, patients taking CYMBALTA did not show a significantly greater pain reduction than patients taking placebo.
In Study OA-1, for various degrees of improvement in pain from baseline to study endpoint, Figure 10 shows the fraction of patients achieving that degree of improvement. The figure is cumulative, so that patients whose change from baseline is, for example, 50%, are also included at every level of improvement below 50%. Patients who did not complete the trial were assigned the value of 0% improvement.
Figure 10: Percentage of Adult Patients with OA Achieving Various Levels of Pain Relief as Measured by 24-Hour Average Pain Severity (Study OA-1)
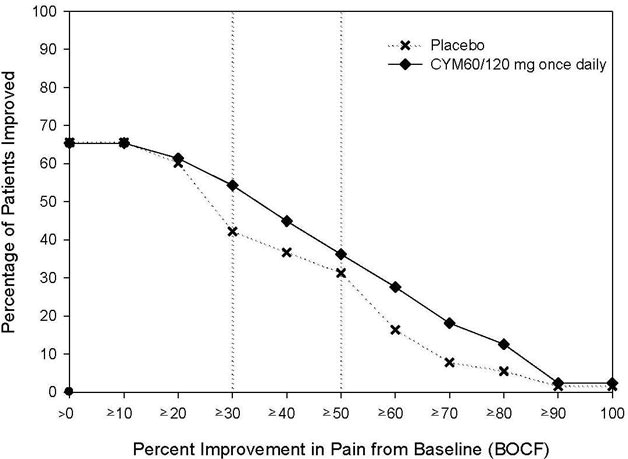
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
CYMBALTA (duloxetine delayed-release capsules) is available in the following strengths, colors, imprints, and presentations:
|
a equivalent to duloxetine base |
|||
| Features | Strengths | ||
| 20 mga | 30 mga | 60 mga | |
| Body color | Opaque green | Opaque white | Opaque green |
| Cap color | Opaque green | Opaque blue | Opaque blue |
| Cap imprint | Lilly 3235 | Lilly 3240 | Lilly 3270 |
| Body imprint | 20mg | 30mg | 60mg |
| Capsule number | PU3235 | PU3240 | PU3270 |
| Presentations and NDC Codes | |||
| Bottles of 30 | NA | 0002-3240-30 | 0002-3270-30 |
| Bottles of 60 | 0002-3235-60 | NA | NA |
| Bottles of 90 | NA | 0002-3240-90 | NA |
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
- Suicidal Thoughts and Behaviors — Advise patients, their families, and their caregivers to look for the emergence of suicidal ideation and behavior, especially during treatment and when the dose is adjusted up or down and instruct them to report such symptoms to their healthcare provider [see Boxed Warning and Warnings and Precautions (5.1)].
- Administration — Advise patients to swallow CYMBALTA whole and to not chew, crush, or open the capsule (do not sprinkle contents on food or mixed with liquids) because these actions might affect the enteric coating.
- Hepatotoxicity — Inform patients that severe liver problems, sometimes fatal, have been reported in patients treated with CYMBALTA. Instruct patients to talk to their healthcare provider if they develop itching, right upper belly pain, dark urine, or yellow skin/eyes while taking CYMBALTA, which may be signs of liver problems. Instruct patients to talk to their healthcare provider about their alcohol consumption. Use of CYMBALTA with heavy alcohol intake may be associated with severe liver injury [see Warnings and Precautions (5.2)].
- Alcohol — Although CYMBALTA does not increase the impairment of mental and motor skills caused by alcohol, use of CYMBALTA concomitantly with heavy alcohol intake may be associated with severe liver injury [see Warnings and Precautions (5.2) and Drug Interactions (7.15)].
- Orthostatic Hypotension, Falls and Syncope — Advise patients of the risk of orthostatic hypotension, falls and syncope, especially during the period of initial use and subsequent dose escalation, and in association with the use of concomitant drugs that might potentiate the orthostatic effect of CYMBALTA [see Warnings and Precautions (5.3)].
- Serotonin Syndrome — Caution patients about the risk of serotonin syndrome with the concomitant use of CYMBALTA and other serotonergic agents including triptans, tricyclic antidepressants, opioids, lithium, buspirone, tryptophan, amphetamines, and St. John's Wort [see Contraindications (4), Warnings and Precautions (5.4), and Drug Interactions (7.14)]. Advise patients of the signs and symptoms associated with serotonin syndrome that may include mental status changes (e.g., agitation, hallucinations, delirium, and coma), autonomic instability (e.g., tachycardia, labile blood pressure, dizziness, diaphoresis, flushing, hyperthermia), neuromuscular changes (e.g., tremor, rigidity, myoclonus, hyperreflexia, incoordination), seizures, and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea). Caution patients to seek medical care immediately if they experience these symptoms.
- Increased Risk of Bleeding — Caution patients about the concomitant use of CYMBALTA and NSAIDs, aspirin, warfarin, or other drugs that affect coagulation since combined use of psychotropic drugs that interfere with serotonin reuptake and these agents has been associated with an increased risk of bleeding [see Warnings and Precautions (5.5) and Use in Specific Populations (8.1)].
- Severe Skin Reactions — Caution patients that CYMBALTA may cause serious skin reactions. This may need to be treated in a hospital and may be life-threatening. Counsel patients to call their doctor right away or get emergency help if they have skin blisters, peeling rash, sores in their mouth, hives, or any other allergic reactions [see Warnings and Precautions (5.6)].
- Discontinuation of Treatment — Instruct patients that discontinuation of CYMBALTA may be associated with symptoms such as dizziness, headache, nausea, diarrhea, paresthesia, irritability, vomiting, insomnia, anxiety, hyperhidrosis, and fatigue, and should be advised not to alter their dosing regimen, or stop taking CYMBALTA without consulting their healthcare provider [see Warnings and Precautions (5.7)].
- Activation of Mania or Hypomania — Adequately screen patients with depressive symptoms for risk of bipolar disorder (e.g. family history of suicide, bipolar disorder, and depression) prior to initiating treatment with CYMBALTA. Advise patients to report any signs or symptoms of a manic reaction such as greatly increased energy, severe trouble sleeping, racing thoughts, reckless behavior, talking more or faster than usual, unusually grand ideas, and excessive happiness or irritability [see Warnings and Precautions (5.8)].
- Angle-Closure Glaucoma — Advise patients that taking CYMBALTA can cause mild pupillary dilation, which in susceptible individuals, can lead to an episode of angle-closure glaucoma. Pre-existing glaucoma is almost always open-angle glaucoma because angle-closure glaucoma, when diagnosed, can be treated definitively with iridectomy. Open-angle glaucoma is not a risk factor for angle-closure glaucoma. Patients may wish to be examined to determine whether they are susceptible to angle-closure, and have a prophylactic procedure (e.g., iridectomy), if they are susceptible [see Warnings and Precautions (5.9)].
- Seizures — Advise patients to inform their healthcare provider if they have a history of seizure disorder [see Warnings and Precautions (5.10)].
- Effects on Blood Pressure — Caution patients that CYMBALTA may cause an increase in blood pressure [see Warnings and Precautions (5.11)].
- Concomitant Medications — Advise patients to inform their healthcare provider if they are taking, or plan to take, any prescription or over-the-counter medications, since there is a potential for interactions [see Dosage and Administration (2.9, 2.10), Contraindications (4), Warnings and Precautions (5.4, 5.12), and Drug Interactions (7)].
- Hyponatremia — Advise patients that hyponatremia has been reported as a result of treatment with SNRIs and SSRIs, including CYMBALTA. Advise patients of the signs and symptoms of hyponatremia [see Warnings and Precautions (5.13)].
- Concomitant Illnesses — Advise patients to inform their healthcare provider about all of their medical conditions [see Warnings and Precautions (5.14)].
- Urinary Hesitation and Retention — CYMBALTA is in a class of medicines that may affect urination. Instruct patients to consult with their healthcare provider if they develop any problems with urine flow [see Warnings and Precautions (5.15)].
- Sexual Dysfunction — Advise patients that use of CYMBALTA may cause symptoms of sexual dysfunction in both male and female patients. Inform patients that they should discuss any changes in sexual function and potential management strategies with their healthcare provider [see Warnings and Precautions (5.16)].
- Pregnancy
- Advise women to notify their healthcare provider if they become pregnant or intend to become pregnant during treatment with CYMBALTA.
- Advise pregnant women or patients who intend to become pregnant that CYMBALTA may increase the risk of neonatal complications requiring prolonged hospitalization, respiratory support, and tube feeding.
- Advise pregnant women that there is a risk of relapse with discontinuation of antidepressants.
- Advise patients that there is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to CYMBALTA during pregnancy [see Use in Specific Populations (8.1)].
- Lactation — Advise breastfeeding women using CYMBALTA to monitor infants for sedation, poor feeding and poor weight gain and to seek medical care if they notice these signs [see Use in Specific Populations (8.2)].
- Interference with Psychomotor Performance — CYMBALTA may be associated with sedation and dizziness. Therefore, caution patients about operating hazardous machinery including automobiles, until they are reasonably certain that CYMBALTA therapy does not affect their ability to engage in such activities.
Marketed by: Lilly USA, LLC, Indianapolis, IN 46285, USA
Copyright © 2004, 2023, Eli Lilly and Company. All rights reserved.
CYM-0014-USPI-20230818
Medication Guide
Cymbalta®
[sim-BALL-tah]
(duloxetine delayed-release capsules)
Read this Medication Guide before you start taking Cymbalta® and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or treatment.
Talk to your healthcare provider about:
- all risks and benefits of treatment with antidepressant medicines
- all treatment choices for depression or other serious mental illness
What is the most important information I should know about antidepressant medicines, depression, other serious mental illnesses, and suicidal thoughts or actions?
1. Cymbalta and other antidepressant medicines may increase suicidal thoughts or actions in some children, teenagers, or young adults within the first few months of treatment or when the dose is changed.
2. Depression and other serious mental illnesses are the most important causes of suicidal thoughts or actions. Some people may have a particularly high risk of having suicidal thoughts or actions. These include people who have (or have a family history of) bipolar illness (also called manic-depressive illness).
3. How can I watch for and try to prevent suicidal thoughts and actions?
- Pay close attention to any changes in mood, behavior, actions, thoughts, or feelings, especially sudden changes. This is very important when an antidepressant medicine is started or when the dose is changed.
- Call your healthcare provider right away to report new or sudden changes in mood, behavior, thoughts, or feelings.
- Keep all follow-up visits with your healthcare provider as scheduled. Call your healthcare provider between visits as needed, especially if you have concerns about symptoms.
Call your healthcare provider right away if you have any of the following symptoms or feelings, especially if they are new, worse, or worry you. In an emergency, call 911.
- attempts to commit suicide
- acting on dangerous impulses
- acting aggressive, being angry, or violent
- thoughts about suicide or dying
- new or worse depression
- new or worse anxiety
- panic attacks
- feeling very agitated or restless
- new or worse irritability
- trouble sleeping
- an extreme increase in activity or talking (mania)
- other unusual changes in behavior or mood
What else do I need to know about antidepressant medicines?
- Never stop an antidepressant medicine without first talking to a healthcare provider. Stopping an antidepressant medicine suddenly can cause other symptoms.
- Antidepressants are medicines used to treat depression and other illnesses. It is important to discuss all the risks of treating depression and also the risks of not treating it. Patients should discuss all treatment choices with your healthcare provider, not just the use of antidepressants.
- Antidepressant medicines have other side effects. Talk to your healthcare provider about the side effects of the medicine prescribed for you or your family member.
- Antidepressant medicines can interact with other medicines. Know all of the medicines that you or your family member takes. Keep a list of all medicines to show your healthcare provider. Do not start new medicines without first checking with your healthcare provider.
What is Cymbalta?
Cymbalta is a prescription medicine used to treat a certain type of depression called Major Depressive Disorder (MDD). Cymbalta belongs to a class of medicines known as SNRIs (or serotonin-norepinephrine reuptake inhibitors).
Cymbalta is also used to treat or manage:
- Generalized Anxiety Disorder (GAD)
- Diabetic Peripheral Neuropathic Pain (DPNP)
- Fibromyalgia (FM)
- Chronic Musculoskeletal Pain
Who should not take Cymbalta?
Do Not take Cymbalta if you:
- take a Monoamine Oxidase Inhibitor (MAOI). Ask your healthcare provider or pharmacist if you are not sure if you take an MAOI, including the antibiotic linezolid or intravenous methylene blue.
- Do not take an MAOI within 5 days of stopping Cymbalta unless directed to do so by your healthcare provider.
- Do not start Cymbalta if you stopped taking an MAOI in the last 14 days unless directed to do so by your healthcare provider.
People who take Cymbalta close in time to an MAOI may have a serious problem called Serotonin Syndrome (see “What are the possible side effects of Cymbalta?”).
What should I tell my healthcare provider before taking Cymbalta?
Before starting Cymbalta, tell your healthcare provider if you:
- have heart problems or high blood pressure
- have diabetes (Cymbalta treatment makes it harder for some people with diabetes to control their blood sugar)
- have liver problems
- have kidney problems
- have glaucoma
- have or had seizures or convulsions
- have bipolar disorder or mania
- have low sodium levels in your blood
- have delayed stomach emptying
- have or had bleeding problems
- are pregnant or plan to become pregnant. Cymbalta may harm your unborn baby. Talk to your healthcare provider about the risk to your unborn baby if you take Cymbalta during pregnancy.
- Tell your healthcare provider right away if you become pregnant or think you are pregnant during treatment with Cymbalta.
- There is a pregnancy registry for females who are exposed to Cymbalta during pregnancy. The purpose of the registry is to collect information about the health of females exposed to Cymbalta and their baby. If you become pregnant during treatment with Cymbalta, talk to your healthcare provider about registering with the National Pregnancy Registry for Antidepressants at 1-866-961-2388 or visit online at
https://womensmentalhealth.org/research/pregnancyregistry/.
- are breastfeeding or plan to breastfeed. Cymbalta passes into your breast milk and may harm your baby. Talk to your healthcare provider about the best way to feed your baby while taking Cymbalta.
Tell your healthcare provider about all the medicines that you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Cymbalta and some medicines may interact with each other, may not work as well, or may cause serious side effects.
Especially tell your healthcare provider if you take:
- triptans used to treat migraine headache
- medicines used to treat mood, anxiety, psychotic or thought disorders, including tricyclics, lithium, buspirone, SSRIs, SNRIs or MAOIs
- tramadol, fentanyl, meperidine, methadone, or other opioids
- amphetamines
- cimetidine
- the antibiotics ciprofloxacin, enoxacin
- medicine to treat irregular heart rate (like propafenone, flecainide, quinidine)
- theophylline
- the blood thinner warfarin (Coumadin, Jantoven)
- non-steroidal anti-inflammatory drug (NSAID) (like ibuprofen, naproxen or aspirin).
- over-the-counter supplements such as tryptophan or St. John’s Wort
- thioridazine (Mellaril). Mellaril together with Cymbalta can cause serious heart rhythm problems or sudden death.
Ask your healthcare provider for a list of these medicines if you are not sure.
Do not take Cymbalta with any other medicine that contain duloxetine.
How should I take Cymbalta?
- Take Cymbalta exactly as your healthcare provider tells you to take it. Your healthcare provider may need to change the dose of Cymbalta until it is the right dose for you.
- Swallow Cymbalta whole. Do not chew or crush Cymbalta.
- Do not open the capsule and sprinkle on food or mix with liquids. Opening the capsule may affect how well Cymbalta works.
- Cymbalta may be taken with or without food.
- If you miss a dose of Cymbalta, take the missed dose as soon as you remember. If it is almost time for the next dose, skip the missed dose and take your next dose at the regular time. Do not take two doses of Cymbalta at the same time.
- If you take too much Cymbalta, call your healthcare provider or poison control center at 1-800-222-1222 right away, or get emergency treatment.
- When switching from another antidepressant to Cymbalta your healthcare provider may want to lower the dose of the initial antidepressant first to potentially avoid side effects.
What should I avoid while taking Cymbalta?
- Cymbalta can cause sleepiness or may affect your ability to make decisions, think clearly, or react quickly. You should not drive, operate heavy machinery, or do other dangerous activities until you know how Cymbalta affects you.
- Use of Cymbalta concomitantly with heavy alcohol intake may be associated with severe liver injury. Avoid heavy alcohol use while taking Cymbalta.
What are the possible side effects of Cymbalta?
Cymbalta may cause serious side effects, including: See “What is the most important information I should know about Cymbalta?”
Common possible side effects in people who take Cymbalta include:
1. liver damage. Symptoms may include:
- itching
- right upper abdominal pain
- dark urine
- yellow skin or eyes
- enlarged liver
- increased liver enzymes
2. changes in blood pressure and falls. Monitor your blood pressure before starting and throughout treatment. Cymbalta may:
- increase your blood pressure.
- decrease your blood pressure when standing and cause dizziness or fainting, mostly when first starting Cymbalta or when increasing the dose.
- increase risk of falls, especially in elderly.
3. Serotonin Syndrome: This condition can be life-threatening and symptoms may include:
- agitation, hallucinations, coma or other changes in mental status
- coordination problems or muscle twitching (overactive reflexes)
- racing heartbeat, high or low blood pressure
- sweating or fever
- nausea, vomiting, or diarrhea
- muscle rigidity
- dizziness
- flushing
- tremor
- seizures
4. abnormal bleeding: Cymbalta and other antidepressant medicines may increase your risk of bleeding or bruising, especially if you take the blood thinner warfarin (Coumadin, Jantoven), a non-steroidal anti-inflammatory drug (NSAIDs, like ibuprofen or naproxen), or aspirin.
5. severe skin reactions: Cymbalta may cause serious skin reactions that may require stopping its use. This may need to be treated in a hospital and may be life-threatening. Call your healthcare provider right away or get emergency help if you have skin blisters, peeling rash, sores in the mouth, hives or any other allergic reactions.
6. discontinuation symptoms: Do not stop Cymbalta without first talking to your healthcare provider. Stopping Cymbalta too quickly or changing from another antidepressant too quickly may result in serious symptoms including:
- anxiety
- irritability
- feeling tired or problems sleeping
- headache
- sweating
- dizziness
- electric shock-like sensations
- vomiting or nausea
- diarrhea
7. manic episodes:
- greatly increased energy
- severe trouble sleeping
- racing thoughts
- reckless behavior
- unusually grand ideas
- excessive happiness or irritability
- talking more or faster than usual
8. visual problems:
- eye pain
- changes in vision
- swelling or redness in or around the eye
Only some people are at risk for these problems. You may want to undergo an eye examination to see if you are at risk and receive preventative treatment if you are.
9. seizures or convulsions
10. low salt (sodium) levels in the blood. Elderly people may be at greater risk for this. Symptoms may include:
- headache
- weakness or feeling unsteady
- confusion, problems concentrating or thinking or memory problems
11. problems with urination. Symptoms may include:
- decreased urine flow
- unable to pass any urine
12. sexual problems (dysfunction). Taking serotonin and norepinephrine reuptake inhibitors (SNRIs), including CYMBALTA, may cause sexual problems.
Symptoms in males may include:
- delayed ejaculation or inability to have an ejaculation
- decreased sex drive
- problems getting or keeping an erection
Symptoms in females may include:
- decreased sex drive
- delayed orgasm or inability to have an orgasm
Talk to your healthcare provider if you develop any changes in your sexual function or if you have any questions or concerns about sexual problems during treatment with CYMBALTA. There may be treatments your healthcare provider can suggest.
The most common side effects of Cymbalta include:
- nausea
- dry mouth
- sleepiness
- fatigue
- constipation
- loss of appetite
- increased sweating
- dizziness
Common possible side effects in children and adolescents who take Cymbalta include:
- nausea
- decreased weight
- dizziness
Side effects in adults may also occur in children and adolescents who take Cymbalta. Children and adolescents should have height and weight monitored during treatment.
Tell your healthcare provider if you have any side effect that bothers you or that does not go away. These are not all the possible side effects of Cymbalta. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to 1-800-FDA-1088.
How should I store Cymbalta?
Store Cymbalta at room temperature between 68°F to 77°F (20°C to 25°C).
Keep Cymbalta and all medicines out of the reach of children.
General information about the safe and effective use of Cymbalta.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use Cymbalta for a condition for which it was not prescribed. Do not give Cymbalta to other people, even if they have the same symptoms that you have. It may harm them.
This Medication Guide summarizes the most important information about Cymbalta. If you would like more information, talk with your healthcare provider. You may ask your healthcare provider or pharmacist for information about Cymbalta that is written for healthcare professionals.
For more information, call 1-800-545-5979.
What are the ingredients in Cymbalta?
Active ingredient: duloxetine hydrochloride
Inactive ingredients:
FD&C Blue No. 2, gelatin, hypromellose, hydroxypropyl methylcellulose acetate succinate, sodium lauryl sulfate, sucrose, sugar spheres, talc, titanium dioxide, and triethyl citrate. The 20 and 60 mg capsules also contain iron oxide yellow.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Coumadin, Jantoven, and Mellaril are trademarks of their respective owners and not trademarks of Eli Lilly and Company.
Medication Guide revised: 08/2023
Marketed by: Lilly USA, LLC
Indianapolis, IN 46285, USA
Cymbalta is a registered trademark of Eli Lilly and Company.
Copyright © 2009, 2023, Eli Lilly and Company. All rights reserved.
CYM-0007-MG-20230818
PACKAGE LABEL- Cymbalta 20 mg, bottle of 60
NDC 0002-3235-60
60 capsules
PU3235
Cymbalta®
duloxetine
delayed release capsules
20 mg
Each capsule contains 22.4 mg of duloxetine hydrochloride equivalent to 20 mg duloxetine.
Rx only
Lilly


