FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
1.1 RET Fusion-Positive Non-Small Cell Lung Cancer
RETEVMO® is indicated for the treatment of adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) with a rearranged during transfection (RET) gene fusion, as detected by an FDA-approved test.
1.2 RET-Mutant Medullary Thyroid Cancer
RETEVMO is indicated for the treatment of adult and pediatric patients 2 years of age and older with advanced or metastatic medullary thyroid cancer (MTC) with a RET mutation, as detected by an FDA-approved test, who require systemic therapy.
1.3 RET Fusion-Positive Thyroid Cancer
RETEVMO is indicated for the treatment of adult and pediatric patients 2 years of age and older with advanced or metastatic thyroid cancer with a RET gene fusion, as detected by an FDA-approved test, who require systemic therapy and who are radioactive iodine-refractory (if radioactive iodine is appropriate).
1.4 Other RET Fusion-Positive Solid Tumors
RETEVMO is indicated for the treatment of adult and pediatric patients 2 years of age and older with locally advanced or metastatic solid tumors with a RET gene fusion, as detected by an FDA-approved test, that have progressed on or following prior systemic treatment or who have no satisfactory alternative treatment options.
This indication is approved under accelerated approval based on overall response rate and duration of response [see Clinical Studies (14.4)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trial(s).
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Select patients for treatment with RETEVMO based on the presence of a RET gene fusion (NSCLC, thyroid cancer, or other solid tumors) or specific RET gene mutation (MTC) in tumor specimens [see Clinical Studies (14)]. Information on FDA-approved test(s) for the detection of RET gene fusions and RET gene mutations is available at: http://www.fda.gov/CompanionDiagnostics. An FDA-approved companion diagnostic test for the detection of RET gene fusions and RET gene mutations in plasma is not available.
2.2 Important Administration Instructions
RETEVMO may be taken with or without food unless coadministered with a proton pump inhibitor (PPI) [see Dosage and Administration (2.4), Clinical Pharmacology (12.3)].
2.3 Recommended Dosage
The recommended dosage of RETEVMO is shown in Table 1:
| Population | RETEVMO Dosage |
| Adult and adolescent patients 12 years of age or older based on body weight | |
|
120 mg twice daily |
|
160 mg twice daily |
| Pediatric patients 2 to less than 12 years of age based on body surface area | |
|
40 mg three times daily |
|
80 mg twice daily |
|
120 mg twice daily |
|
160 mg twice daily |
| Dosing pediatric patients with body surface area less than 0.33 m2 is not recommended | |
Continue treatment with RETEVMO until disease progression or unacceptable toxicity.
Swallow the capsules whole. Do not crush or chew the capsules. Do not administer to pediatric patients who are unable to swallow a capsule.
Swallow the tablets whole. Do not crush or chew the tablets.
Do not take a missed dose unless it is more than 6 hours until next scheduled dose.
If vomiting occurs after RETEVMO administration, do not take an additional dose and continue to the next scheduled time for the next dose.
2.4 Dosage Modifications for Concomitant Use of Acid-Reducing Agents
Avoid concomitant use of a PPI, a histamine-2 (H2) receptor antagonist, or a locally-acting antacid with RETEVMO [see Drug Interactions (7.1)]. If concomitant use cannot be avoided:
- Take RETEVMO with food when coadministered with a PPI.
- Take RETEVMO 2 hours before or 10 hours after administration of an H2 receptor antagonist.
- Take RETEVMO 2 hours before or 2 hours after administration of a locally-acting antacid.
2.5 Dosage Modifications for Adverse Reactions
The recommended dose reductions for adverse reactions are provided in Table 2.
| Current RETEVMO Dosage |
Dose Reduction | ||
| First | Second | Third | |
| 40 mg three times daily | 40 mg twice daily | 40 mg once daily | permanently discontinue |
| 80 mg twice daily | 40 mg twice daily | 40 mg once daily | permanently discontinue |
| 120 mg twice daily | 80 mg twice daily | 40 mg twice daily | 40 mg once daily |
| 160 mg twice daily | 120 mg twice daily | 80 mg twice daily | 40 mg twice daily |
| Permanently discontinue RETEVMO in patients unable to tolerate three dose reductions. | |||
The recommended dosage modifications for adverse reactions are provided in Table 3.
| Adverse Reaction | Severity | Dosage Modification |
| Hepatotoxicity [see Warnings and Precautions (5.1)] |
Grade 3 or Grade 4 |
|
| Interstitial Lung Disease/ Pneumonitis [see Warnings and Precautions (5.2)] |
Grade 2 |
|
| Grade 3 or Grade 4 |
|
|
| Hypertension [see Warnings and Precautions (5.3)] |
Grade 3 |
|
| Grade 4 |
|
|
| QT Interval Prolongation [see Warnings and Precautions (5.4)] |
Grade 3 |
|
| Grade 4 |
|
|
| Hemorrhagic Events [see Warnings and Precautions (5.5)] |
Grade 3 or Grade 4 |
|
| Hypersensitivity Reactions [see Warnings and Precautions (5.6)] |
All Grades |
|
| Hypothyroidism [see Warnings and Precautions (5.9)] |
Grade 3 or Grade 4 |
|
| Other Adverse Reactions [see Adverse Reactions (6.1)] |
Grade 3 or Grade 4 |
|
2.6 Dosage Modifications for Concomitant Use of Strong and Moderate CYP3A Inhibitors
Avoid concomitant use of strong and moderate CYP3A inhibitors with RETEVMO. If concomitant use of a strong or moderate CYP3A inhibitor cannot be avoided, reduce the RETEVMO dose as recommended in Table 4. After the inhibitor has been discontinued for 3 to 5 elimination half-lives, resume RETEVMO at the dose taken prior to initiating the CYP3A inhibitor [see Drug Interactions (7.1)].
| Current RETEVMO Dosage | Recommended RETEVMO Dosage | |
| Moderate CYP3A Inhibitor | Strong CYP3A Inhibitor | |
| 40 mg orally three times daily | 40 mg orally once daily | 40 mg orally once daily |
| 80 mg orally twice daily | 40 mg orally twice daily | 40 mg orally twice daily |
| 120 mg orally twice daily | 80 mg orally twice daily | 40 mg orally twice daily |
| 160 mg orally twice daily | 120 mg orally twice daily | 80 mg orally twice daily |
2.7 Dosage Modification for Severe Hepatic Impairment
Reduce the recommended dosage of RETEVMO for patients with severe hepatic impairment as recommended in Table 5[see Use in Specific Populations (8.7)].
| Current RETEVMO Dosage | Recommended RETEVMO Dosage |
| 40 mg orally three times daily | 40 mg orally twice daily |
| 80 mg orally twice daily | 40 mg orally twice daily |
| 120 mg orally twice daily | 80 mg orally twice daily |
| 160 mg orally twice daily | 80 mg orally twice daily |
3 DOSAGE FORMS AND STRENGTHS
Capsules:
- 40 mg: gray opaque capsule imprinted with “Lilly”, “3977” and “40 mg” in black ink.
- 80 mg: blue opaque capsule imprinted with “Lilly”, “2980” and “80 mg” in black ink.
Tablets:
- 40 mg: light gray, film coated, round tablet debossed with “Ret 40” on one side and “5340” on the other side.
- 80 mg: dark red-purple, film coated, round tablet debossed with “Ret 80” on one side and “6082” on the other side.
- 120 mg: light purple, film coated, round tablet debossed with “Ret 120” on one side and “6120” on the other side.
- 160 mg: light pink, film coated, round tablet debossed with “Ret 160” on one side and “5562” on the other side.
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
Serious hepatic adverse reactions occurred in 3% of patients treated with RETEVMO. Increased AST occurred in 59% of patients, including Grade 3 or 4 events in 11% and increased ALT occurred in 55% of patients, including Grade 3 or 4 events in 12% [see Adverse Reactions (6.1)]. The median time to first onset for increased AST was 6 weeks (range: 1 day to 3.4 years) and increased ALT was 5.8 weeks (range: 1 day to 2.5 years).
Monitor ALT and AST prior to initiating RETEVMO, every 2 weeks during the first 3 months, then monthly thereafter and as clinically indicated. Withhold, reduce the dose or permanently discontinue RETEVMO based on the severity [see Dosage and Administration (2.5)].
5.2 Interstitial Lung Disease/Pneumonitis
Severe, life-threatening, and fatal interstitial lung disease (ILD)/pneumonitis can occur in patients treated with RETEVMO. ILD/pneumonitis occurred in 1.8% of patients who received RETEVMO, including 0.3% with Grade 3 or 4 events, and 0.3% with fatal reactions.
Monitor for pulmonary symptoms indicative of ILD/pneumonitis. Withhold RETEVMO and promptly investigate for ILD in any patient who presents with acute or worsening of respiratory symptoms which may be indicative of ILD (e.g., dyspnea, cough, and fever). Withhold, reduce the dose or permanently discontinue RETEVMO based on severity of confirmed ILD [see Dosage and Administration (2.5)].
5.3 Hypertension
Hypertension occurred in 41% of patients, including Grade 3 hypertension in 20% and Grade 4 in one (0.1%) patient [see Adverse Reactions (6.1)]. Overall, 6.3% had their dose interrupted and 1.3% had their dose reduced for hypertension. Treatment-emergent hypertension was most commonly managed with anti-hypertension medications.
Do not initiate RETEVMO in patients with uncontrolled hypertension. Optimize blood pressure prior to initiating RETEVMO. Monitor blood pressure after 1 week, at least monthly thereafter and as clinically indicated. Initiate or adjust anti-hypertensive therapy as appropriate. Withhold, reduce the dose, or permanently discontinue RETEVMO based on the severity [see Dosage and Administration (2.5)].
5.4 QT Interval Prolongation
RETEVMO can cause concentration-dependent QT interval prolongation [see Clinical Pharmacology (12.2)]. An increase in QTcF interval to >500 ms was measured in 7% of patients and an increase in the QTcF interval of at least 60 ms over baseline was measured in 20% of patients [see Adverse Reactions (6.1)]. RETEVMO has not been studied in patients with clinically significant active cardiovascular disease or recent myocardial infarction.
Monitor patients who are at significant risk of developing QTc prolongation, including patients with known long QT syndromes, clinically significant bradyarrhythmias, and severe or uncontrolled heart failure. Assess QT interval, electrolytes and TSH at baseline and periodically during treatment, adjusting frequency based upon risk factors including diarrhea. Correct hypokalemia, hypomagnesemia and hypocalcemia prior to initiating RETEVMO and during treatment.
Monitor the QT interval more frequently when RETEVMO is concomitantly administered with strong and moderate CYP3A inhibitors or drugs known to prolong QTc interval. Withhold and reduce the dose or permanently discontinue RETEVMO based on the severity [see Dosage and Administration (2.5)].
5.5 Hemorrhagic Events
Serious including fatal hemorrhagic events can occur with RETEVMO. Grade ≥3 hemorrhagic events occurred in 3.1% of patients treated with RETEVMO, including 4 (0.5%) patients with fatal hemorrhagic events, including cerebral hemorrhage (n = 2), tracheostomy site hemorrhage (n = 1), and hemoptysis (n=1).
Permanently discontinue RETEVMO in patients with severe or life-threatening hemorrhage [see Dosage and Administration (2.5)].
5.6 Hypersensitivity
Hypersensitivity occurred in 6% of patients receiving RETEVMO, including Grade 3 hypersensitivity in 1.9%. The median time to onset was 1.9 weeks (range: 5 days to 2 years). Signs and symptoms of hypersensitivity included fever, rash and arthralgias or myalgias with concurrent decreased platelets or transaminitis.
If hypersensitivity occurs, withhold RETEVMO and begin corticosteroids at a dose of 1 mg/kg prednisone (or equivalent). Upon resolution of the event, resume RETEVMO at a reduced dose and increase the dose of RETEVMO by 1 dose level each week as tolerated until reaching the dose taken prior to onset of hypersensitivity [see Dosage and Administration (2.5)]. Continue steroids until patient reaches target dose and then taper. Permanently discontinue RETEVMO for recurrent hypersensitivity.
5.7 Tumor Lysis Syndrome
Tumor lysis syndrome (TLS) occurred in 0.6% of patients with medullary thyroid carcinoma receiving RETEVMO [see Adverse Reactions (6.1)]. Patients may be at risk of TLS if they have rapidly growing tumors, a high tumor burden, renal dysfunction, or dehydration. Closely monitor patients at risk, consider appropriate prophylaxis including hydration, and treat as clinically indicated.
5.8 Risk of Impaired Wound Healing
Impaired wound healing can occur in patients who receive drugs that inhibit the vascular endothelial growth factor (VEGF) signaling pathway. Therefore, RETEVMO has the potential to adversely affect wound healing.
Withhold RETEVMO for at least 7 days prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of RETEVMO after resolution of wound healing complications has not been established.
5.9 Hypothyroidism
RETEVMO can cause hypothyroidism. Hypothyroidism occurred in 13% of patients treated with RETEVMO; all reactions were Grade 1 or 2. Hypothyroidism occurred in 13% of patients (50/373) with thyroid cancer and 13% of patients (53/423) with other solid tumors including NSCLC [see Adverse Reactions (6.1)].
Monitor thyroid function before treatment with RETEVMO and periodically during treatment. Treat with thyroid hormone replacement as clinically indicated. Withhold RETEVMO until clinically stable or permanently discontinue RETEVMO based on severity [see Dosage and Administration (2.5)].
5.10 Embryo-Fetal Toxicity
Based on data from animal reproduction studies and its mechanism of action, RETEVMO can cause fetal harm when administered to a pregnant woman. Administration of selpercatinib to pregnant rats during organogenesis at maternal exposures that were approximately equal to those observed at the recommended human dose of 160 mg twice daily resulted in embryolethality and malformations.
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with RETEVMO and for 1 week after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with RETEVMO and for 1 week after the last dose [see Use in Specific Populations (8.1, 8.3)].
5.11 Slipped Capital Femoral Epiphysis/Slipped Upper Femoral Epiphysis in Pediatric Patients
Slipped capital femoral epiphysis/slipped upper femoral epiphysis (SCFE/SUFE) occurred in 1 adolescent (3.7% of 27 patients) receiving RETEVMO in LIBRETTO-121 and 1 adolescent (0.5% of 193 patients) receiving RETEVMO in LIBRETTO-531 [see Adverse Reactions (6.1)]. Monitor patients for symptoms indicative of SCFE/SUFE and treat as medically and surgically appropriate [see Adverse Reactions (6.1)].
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Hepatotoxicity [see Warnings and Precautions (5.1)]
- Interstitial Lung Disease / Pneumonitis [see Warnings and Precautions (5.2)]
- Hypertension [see Warnings and Precautions (5.3)]
- QT Interval Prolongation [see Warnings and Precautions (5.4)]
- Hemorrhagic Events [see Warnings and Precautions (5.5)]
- Hypersensitivity [see Warnings and Precautions (5.6)]
- Tumor Lysis Syndrome [see Warnings and Precautions (5.7)]
- Risk of Impaired Wound Healing [see Warnings and Precautions (5.8)]
- Hypothyroidism [see Warnings and Precautions (5.9)]
- Slipped Capital Femoral Epiphysis/Slipped Upper Femoral Epiphysis in Adolescent Patients [see Warnings and Precautions (5.11)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety population described in the WARNINGS and PRECAUTIONS and below reflects exposure to RETEVMO as a single agent administered at 160 mg orally twice daily evaluated in 796 patients with advanced solid tumors in LIBRETTO-001 [see Clinical Studies (14)].
RETGene Fusion or Gene Mutation Positive Solid Tumors
LIBRETTO-001
Among the 796 patients who received RETEVMO, 84% were exposed for 6 months or longer and 73% were exposed for greater than one year. Among these patients, 96% received at least one dose of RETEVMO at the recommended dosage of 160 mg orally twice daily.
The median age was 59 years (range: 15 to 92 years); 0.3% were pediatric patients 12 to 16 years of age; 51% were male; and 69% were White, 23% were Asian, and 3% were Black or African American; and 5% were Hispanic/Latino. The most common tumors were NSCLC (45%), MTC (40%), and non-medullary thyroid carcinoma (7%).
Serious adverse reactions occurred in 44% of patients who received RETEVMO. The most frequent serious adverse reactions (≥2% of patients) were pneumonia, pleural effusion, abdominal pain, hemorrhage, hypersensitivity, dyspnea, and hyponatremia. Fatal adverse reactions occurred in 3% of patients; fatal adverse reactions included sepsis (n = 6), respiratory failure (n = 5), hemorrhage (n = 4), pneumonia (n = 3), pneumonitis (n = 2), cardiac arrest (n=2), sudden death (n = 1), and cardiac failure (n = 1).
Permanent discontinuation due to an adverse reaction occurred in 8% of patients who received RETEVMO. Adverse reactions resulting in permanent discontinuation in ≥0.5% of patients included increased ALT (0.6%), fatigue (0.6%), sepsis (0.5%), and increased AST (0.5%).
Dosage interruptions due to an adverse reaction occurred in 64% of patients who received RETEVMO. Adverse reactions requiring dosage interruption in ≥5% of patients included increased ALT, increased AST, diarrhea, and hypertension.
Dose reductions due to an adverse reaction occurred in 41% of patients who received RETEVMO. Adverse reactions requiring dosage reductions in ≥2% of patients included increased ALT, increased AST, QT prolongation, fatigue, diarrhea, drug hypersensitivity, and edema.
The most common adverse reactions (≥25%) were edema, diarrhea, fatigue, dry mouth, hypertension, abdominal pain, constipation, rash, nausea, and headache.
The most common Grade 3 or 4 laboratory abnormalities (≥5%) were decreased lymphocytes, increased alanine aminotransferase (ALT), increased aspartate aminotransferase (AST), decreased sodium, and decreased calcium.
Table 6 summarizes the adverse reactions in LIBRETTO-001.
|
1 Edema includes edema peripheral, face edema, periorbital edema, eye edema, eyelid edema, orbital edema, localized edema, lymphedema, scrotal edema, peripheral swelling, scrotal swelling, swelling, swelling face, eye swelling, generalized edema, genital edema. |
||
|
2 Fatigue includes asthenia and malaise. |
||
|
3 Diarrhea includes defecation urgency, frequent bowel movements, gastrointestinal hypermotility, anal incontinence. |
||
|
4 Abdominal pain includes abdominal pain upper, abdominal pain lower, abdominal discomfort, abdominal tenderness, epigastric discomfort, gastrointestinal pain. |
||
|
5 Rash includes rash erythematous, rash macular, rash maculopapular, rash morbilliform, rash papular, rash pruritic, butterfly rash, exfoliative rash, rash follicular, rash generalized, rash vesicular. |
||
|
6 Headache includes sinus headache, tension headache. |
||
|
7 Cough includes productive cough, upper airway cough syndrome. |
||
|
8 Dyspnea includes dyspnea exertional, dyspnea at rest. |
||
|
9 Hemorrhage includes, epistaxis, hematuria, hemoptysis, contusion, rectal hemorrhage, vaginal hemorrhage, ecchymosis, hematochezia, petechiae, traumatic hematoma, anal hemorrhage, blood blister, blood urine present, cerebral hemorrhage, gastric hemorrhage, hemorrhage intracranial, hemorrhage subcutaneous, spontaneous hematoma, abdominal wall hematoma, angina bullosa hemorrhagica, conjunctival hemorrhage, disseminated intravascular coagulation, diverticulum intestinal hemorrhagic, eye hemorrhage, gastrointestinal hemorrhage, gingival bleeding, hematemesis, hemorrhagic stroke, hemorrhoidal hemorrhage, hepatic hemorrhage, hepatic hematoma, intraabdominal hemorrhage, laryngeal hemorrhage, lower gastrointestinal hemorrhage, melena, mouth hemorrhage, occult blood positive, post procedural hemorrhage, postmenopausal hemorrhage, pelvic hematoma, periorbital hematoma, periorbital hemorrhage, pharyngeal hemorrhage, pulmonary contusion, purpura, retinal hemorrhage, retroperitoneal hematoma, scleral hemorrhage, skin hemorrhage, subarachnoid hemorrhage, subdural hemorrhage, upper gastrointestinal hemorrhage, uterine hemorrhage, vessel puncture site hematoma. |
||
|
* Only includes a grade 3 adverse reaction. |
||
|
# Graded according to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 4.03 |
||
| Adverse Reaction | RETEVMO (n = 796) |
|
| Grades 1-4# (%) |
Grades 3-4 (%) |
|
| General Disorders and Administration Site Conditions | ||
| Edema1 | 49 | 0.8* |
| Fatigue2 | 46 | 3.1* |
| Arthralgia | 21 | 0.3* |
| Gastrointestinal Disorders | ||
| Diarrhea3 | 47 | 5* |
| Dry Mouth | 43 | 0 |
| Abdominal pain4 | 34 | 2.5* |
| Constipation | 33 | 0.8* |
| Nausea | 31 | 1.1* |
| Vomiting | 22 | 1.8* |
| Vascular Disorders | ||
| Hypertension | 41 | 20 |
| Skin and Subcutaneous Tissue Disorders | ||
| Rash5 | 33 | 0.6* |
| Nervous System Disorders | ||
| Headache6 | 28 | 1.4* |
| Respiratory, Thoracic and Mediastinal Disorders | ||
| Cough7 | 24 | 0 |
| Dyspnea8 | 22 | 3.1 |
| Blood and Lymphatic System Disorders | ||
| Hemorrhage9 | 22 | 2.6 |
| Investigations | ||
| Prolonged QT interval | 21 | 4.8* |
Clinically relevant adverse reactions in ≤15% of patients who received RETEVMO include hypothyroidism (13%); pneumonia (11%), hypersensitivity (6%); interstitial lung disease/pneumonitis, chylothorax, chylous ascites or tumor lysis syndrome (all < 2%).
Table 7 summarizes the laboratory abnormalities in LIBRETTO-001.
|
1 Denominator for each laboratory parameter is based on the number of patients with a baseline and post-treatment laboratory value available, which ranged from 765 to 791 patients. |
||
|
# Graded according to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 4.03 |
||
| Laboratory Abnormality | RETEVMO1 | |
| Grades 1-4# (%) |
Grades 3-4 (%) |
|
| Chemistry | ||
| Increased AST | 59 | 11 |
| Decreased calcium | 59 | 5.7 |
| Increased ALT | 56 | 12 |
| Decreased albumin | 56 | 2.3 |
| Increased glucose | 53 | 2.8 |
| Increased creatinine | 47 | 2.4 |
| Decreased sodium | 42 | 11 |
| Increased alkaline phosphatase | 40 | 3.4 |
| Increased total cholesterol | 35 | 1.7 |
| Increased potassium | 34 | 2.7 |
| Decreased glucose | 34 | 1.0 |
| Decreased magnesium | 33 | 0.6 |
| Increased bilirubin | 30 | 2.8 |
| Hematology | ||
| Decreased lymphocytes | 52 | 20 |
| Decreased platelets | 37 | 3.2 |
| Decreased hemoglobin | 28 | 3.5 |
| Decreased neutrophils | 25 | 3.2 |
LIBRETTO-121
The safety population described below reflects exposure to RETEVMO as a single agent at 92 mg/m2 orally twice daily evaluated in 27 patients with advanced solid tumors harboring an activating RET alteration in LIBRETTO-121 [see Clinical Studies (14)]. Among the 27 pediatric and adolescent patients who received RETEVMO, 81% were exposed for 6 months or longer and 59% were exposed for greater than one year.
The median age was 13 years (range: 2 to 20 years); 22% were pediatric patients 2 to 12 years of age; 59% were male; and 52% were White, 26% were Asian, and 11% were Black or African American; and 19% were Hispanic/Latino. The most common cancers were MTC (52%), and papillary thyroid cancer (37%).
Serious adverse reactions occurred in 22% of patients who received RETEVMO. The serious adverse reactions (in 1 patient each) were abdominal infection, abdominal pain, aspiration, constipation, diarrhea, epiphysiolysis, nausea, pneumonia, pneumatosis intestinalis, rhinovirus infection, sepsis, vomiting.
Dosage interruptions due to an adverse reaction occurred in 22% of patients who received RETEVMO. Adverse reactions requiring dosage interruption in ≥5% of patients included decreased neutrophils.
Dose reductions due to an adverse reaction occurred in 15% of patients who received RETEVMO. Adverse reactions requiring dosage reductions in ≥2% of patients included decreased neutrophils, increased ALT, and increased weight.
The most common adverse reactions (≥25%) were musculoskeletal pain, diarrhea, headache, nausea, vomiting, coronavirus infection, abdominal pain, fatigue, pyrexia, and hemorrhage.
The most common Grade 3 or 4 laboratory abnormalities (≥5%) were decreased calcium, decreased hemoglobin, and decreased neutrophils.
Table 8 summarizes the adverse reactions in LIBRETTO-121.
| Adverse Reactions | RETEVMO N= 27 |
|
|---|---|---|
| Grades 1-4# % |
Grades 3-4 % |
|
|
1 Musculoskeletal pain includes arthralgia, back pain, bone pain, musculoskeletal chest pain, non-cardiac chest pain, neck pain, pain in extremity |
||
|
2 Diarrhea includes anal incontinence |
||
|
3 Abdominal pain includes abdominal pain upper |
||
|
4 Stomatitis includes angular cheilitis |
||
|
5 Fatigue includes asthenia and malaise |
||
|
6 Edema includes edema peripheral, face edema, localized edema, generalized edema, swelling |
||
|
7 Hemorrhage includes mouth hemorrhage, epistaxis |
||
|
8 Hypothyroidism includes blood thyroid stimulating hormone increased, thyroglobulin increased |
||
|
9 Rash includes rash maculopapular |
||
|
* No Grade 4 events were reported. |
||
|
# Graded according to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 5.0. |
||
| Musculoskeletal and Connective Tissue Disorders | ||
| Musculoskeletal pain1 | 56 | 0 |
| Gastrointestinal disorders | ||
| Diarrhea2 | 41 | 0 |
| Nausea | 30 | 3.7* |
| Vomiting | 30 | 7* |
| Abdominal pain3 | 26 | 0 |
| Constipation | 19 | 7* |
| Stomatitis4 | 15 | 0 |
| Nervous System Disorders | ||
| Headache | 33 | 0 |
| Infections and Infestations | ||
| Coronavirus infection | 30 | 0 |
| Upper respiratory tract infection | 22 | 0 |
| General Disorders and Administration Site Conditions | ||
| Fatigue5 | 26 | 0 |
| Pyrexia | 26 | 0 |
| Edema6 | 19 | 0 |
| Increased weight | 19 | 7* |
| Blood and Lymphatic System Disorders | ||
| Hemorrhage7 | 26 | 3.7* |
| Respiratory, Thoracic and Mediastinal Disorders | ||
| Oropharyngeal pain | 22 | 0 |
| Cough | 22 | 0 |
| Endocrine Disorders | ||
| Hypothyroidism8 | 19 | 0 |
| Skin and Subcutaneous Tissue Disorders | ||
| Rash9 | 19 | 0 |
| Renal and Urinary Disorders | ||
| Proteinuria | 15 | 0 |
Clinically relevant adverse reactions in <15% of patients who received RETEVMO include dizziness (11%), urinary tract infection (11%), decreased appetite (7%), electrocardiogram QT prolonged (7%), hypersensitivity (7%), hypertension (7%), and pneumonia (3.7%).
Table 9 summarizes the laboratory abnormalities in LIBRETTO-121.
|
1 Denominator for each laboratory parameter is based on the number of patients with a baseline and post-treatment laboratory value available, which ranged from 21 to 27 patients. |
||
|
* No Grade 4 abnormalities were reported. |
||
|
# Graded according to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 5. |
||
| Laboratory Abnormality | RETEVMO1 | |
| Grades 1-4# (%) |
Grades 3-4 (%) |
|
| Chemistry | ||
| Decreased calcium | 59 | 7 |
| Increased ALT | 56 | 3.7* |
| Increased alkaline phosphatase | 52 | 0 |
| Increased AST | 48 | 3.7* |
| Decreased albumin | 44 | 0 |
| Increased bilirubin | 30 | 0 |
| Increased creatinine | 22 | 0 |
| Decreased potassium | 22 | 3.7 |
| Decreased magnesium | 15 | 3.7 |
| Hematology | ||
| Decreased neutrophils | 44 | 7* |
| Decreased lymphocytes | 24 | 4.8 |
| Decreased platelets | 22 | 0 |
| Decreased hemoglobin | 19 | 7* |
Treatment-naïve RET Fusion-Positive Non-small Cell Lung Cancer
LIBRETTO-431
The safety population described below reflects exposure to RETEVMO as a single agent administered at 160 mg orally twice daily evaluated in 158 patients with unresectable locally advanced or metastatic RET fusion-positive NSCLC in LIBRETTO-431 [see Clinical Studies (14)]. Among the 158 patients who received RETEVMO, the median duration of exposure was 16.7 months (range: 5 days to 37.9 months); 87% were exposed for 6 months or longer and 70% were exposed for one year or longer.
The median age was 61 years (range: 31 to 87 years); 46% were male; and 36% were White, 58% were Asian, 1.3% were Black or African American, 1.3% were American Indian or Alaska Native, and 3.2% were missing.
Serious adverse reactions occurred in 35% of patients who received RETEVMO. The most frequent serious adverse reactions (≥2% of patients) were pleural effusion, and abnormal hepatic function. Fatal adverse reactions occurred in 4.4% of patients who received RETEVMO; fatal adverse reactions included myocardial infarction (n = 2), respiratory failure (n = 2), cardiac arrest, malnutrition, and sudden death (n = 1, each).
Permanent discontinuation due to an adverse reaction occurred in 10% of patients who received RETEVMO. Adverse reactions resulting in permanent discontinuation in ≥1% of patients included increased ALT (1.3%), and myocardial infarction (1.3%).
Dosage interruptions due to an adverse reaction occurred in 72% of patients who received RETEVMO. Adverse reactions requiring dosage interruption in ≥5% of patients included increased ALT, hypertension, increased AST, QT prolongation, diarrhea, and COVID-19 infection.
Dose reductions due to an adverse reaction occurred in 51% of patients who received RETEVMO. Adverse reactions requiring dose reductions in ≥5% of patients included increased ALT, increased AST, QT prolongation.
The most common adverse reactions (≥25%) in patients who received RETEVMO were hypertension, diarrhea, edema, dry mouth, rash, fatigue, abdominal pain, and musculoskeletal pain.
The most common Grade 3 or 4 laboratory abnormalities (≥5%) in patients who received RETEVMO were increased ALT, increased AST, and decreased lymphocytes.
Table 10 summarizes the adverse reactions in LIBRETTO-431.
|
1 Diarrhea includes diarrhea, anal incontinence. |
||||
|
2 Dry mouth includes dry mouth, mucosal dryness. |
||||
|
3 Abdominal pain includes abdominal pain, abdominal pain upper, abdominal discomfort, abdominal pain lower, gastrointestinal pain. |
||||
|
4 Stomatitis includes stomatitis, mouth ulceration, mucosal inflammation. |
||||
|
5 Vomiting includes vomiting, retching, regurgitation. |
||||
|
6 Edema includes edema, edema peripheral, face edema, periorbital edema, swelling face, peripheral swelling, localized edema, eyelid edema, orbital edema, eye edema, scrotal edema, penile edema, orbital swelling, periorbital swelling. |
||||
|
7 Fatigue includes fatigue, asthenia, malaise. |
||||
|
8 Rash includes rash, rash maculopapular, skin exfoliation, rash erythematous, rash macular, dermatitis, urticaria, rash papular, dermatitis allergic, rash pustular, rash vesicular, genital rash. |
||||
|
9 Musculoskeletal pain includes musculoskeletal pain, arthralgia, back pain, bone pain, musculoskeletal chest pain, non-cardiac chest pain, neck pain, pain in extremity. |
||||
|
* No Grade 4 abnormalities were reported. |
||||
|
# Graded according to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 5.0. |
||||
| Adverse Reaction | RETEVMO (n=158) |
Chemotherapy with or without pembrolizumab (n=98) |
||
| Grades 1-4# (%) |
Grades 3-4 (%) |
Grades 1-4# (%) |
Grades 3-4 (%) |
|
| Vascular disorders | ||||
| Hypertension | 48 | 20* | 7 | 3.1* |
| Gastrointestinal disorders | ||||
| Diarrhea1 | 44 | 1.3* | 24 | 2.0* |
| Dry mouth2 | 39 | 0 | 6 | 0 |
| Abdominal pain3 | 25 | 0.6* | 19 | 2.0* |
| Constipation | 22 | 0 | 40 | 1.0* |
| Stomatitis4 | 18 | 0 | 16 | 0 |
| Nausea | 13 | 0 | 44 | 1.0* |
| Vomiting5 | 13 | 0 | 23 | 1.0* |
| General disorders and administration site conditions | ||||
| Edema6 | 41 | 2.5* | 28 | 0 |
| Fatigue7 | 32 | 3.2* | 50 | 5* |
| Pyrexia | 13 | 0.6* | 23 | 0 |
| Skin and subcutaneous tissue disorders | ||||
| Rash8 | 33 | 1.9* | 30 | 1.0* |
| Musculoskeletal and Connective Tissue Disorders | ||||
| Musculoskeletal pain9 | 25 | 0 | 28 | 0 |
| Investigations | ||||
| Electrocardiogram QT prolonged | 20 | 9* | 1.0 | 0 |
| Infections and infestations | ||||
| COVID-19 infection | 19 | 0.6* | 18 | 0 |
| Metabolism and nutrition disorders | ||||
| Decreased appetite | 17 | 0 | 34 | 2.0* |
Clinically relevant adverse reactions in <15% of patients who received RETEVMO include headache (14%); hemorrhage (13%); urinary tract infections (12%); hypothyroidism (9%); pneumonia (9%); dizziness (8%); interstitial lung disease/pneumonitis (4.4%); hypersensitivity, chylous ascites, and chylothorax (all < 2%).
Table 11 summarizes the laboratory abnormalities in LIBRETTO-431.
|
1 Denominator for each laboratory parameter is based on the number of patients with a baseline and post-treatment laboratory value available: RETEVMO (range: 154 to 157 patients) and chemotherapy with or without pembrolizumab (range: 96 to 97 patients). |
||||
|
# Graded according to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 5.0. |
||||
| Laboratory Abnormality1 | RETEVMO | Chemotherapy with or without pembrolizumab | ||
| Grades 1-4# (%) |
Grades 3-4 (%) |
Grades 1-4# (%) |
Grades 3-4 (%) |
|
| Chemistry | ||||
| ALT increased | 81 | 21 | 63 | 4.1 |
| AST increased | 77 | 10 | 46 | 0 |
| Alkaline phosphatase Increased | 35 | 1.3 | 22 | 0 |
| Total bilirubin Increased | 52 | 1.3 | 9 | 0 |
| Blood creatinine Increased | 23 | 0 | 21 | 0 |
| Magnesium decreased | 16 | 0.6 | 8 | 0 |
| Albumin decreased | 25 | 0 | 5 | 0 |
| Calcium decreased | 53 | 1.9 | 24 | 1.0 |
| Sodium decreased | 31 | 3.2 | 41 | 2.1 |
| Potassium decreased | 17 | 1.3 | 15 | 1.0 |
| Hematology | ||||
| Platelets decreased | 53 | 3.2 | 39 | 5 |
| Lymphocyte count decreased | 53 | 8 | 64 | 15 |
| Hemoglobin decreased | 21 | 0 | 91 | 5 |
| Neutrophil count decreased | 53 | 2.0 | 58 | 11 |
Increased Creatinine
In healthy subjects administered RETEVMO 160 mg orally twice daily, serum creatinine increased 18% after 10 days. Consider alternative markers of renal function if persistent elevations in serum creatinine are observed [see Clinical Pharmacology (12.3)].
RET-Mutant Medullary Thyroid Cancer
LIBRETTO-531
The safety population described below reflects exposure to RETEVMO as a single agent administered at 160 mg (adults) or at 92 mg/m2 (adolescent, not to exceed 160 mg) orally twice daily, in patients with progressive, advanced, kinase inhibitor naïve, RET-mutant medullary thyroid cancer in LIBRETTO-531 [see Clinical Studies (14.2)]. Among the 193 patients who received RETEVMO, the observed median duration of exposure was 14.5 months (range: 25 days to 36 months); 80% were exposed for 6 months or longer and 59% were exposed for one year or longer.
The median age was 55 years (range: 12 to 84 years); 63% were male; and 69% were White, 28% were Asian, 2.9% were Black or African American and ethnicity was not routinely collected.
Serious adverse reactions occurred in 22% of patients who received RETEVMO. The most frequent serious adverse reactions were pneumonia and pyrexia (n = 3, each) and hypertension and urinary tract infection (n = 2, each). Fatal adverse reactions occurred in 2.1% of patients; fatal adverse reactions included COVID-19, diabetic ketoacidosis, multiple organ dysfunction syndrome, and sudden death (n=1 each).
Permanent discontinuation due to an adverse reaction occurred in 4.7% of patients who received RETEVMO. Adverse reactions resulting in permanent discontinuation were edema, multiple organ dysfunction syndrome, sudden death, AST increased, diabetic ketoacidosis, chronic kidney disease, retinopathy, COVID-19, and somatic symptom disorder (n = 1, each).
Dosage interruptions due to an adverse reaction occurred in 49% of patients who received RETEVMO. Adverse reactions requiring dosage omission in ≥5% of patients included ALT increased (9%) and hypertension (7%).
Dose reductions due to an adverse reaction occurred in 39% of patients who received RETEVMO. One adverse reaction, increased ALT (7%), required a dose reduction in ≥5% of patients.
The most common adverse reactions (≥25%) in patients who received RETEVMO were hypertension, edema, dry mouth, fatigue, and diarrhea.
The most common Grade 3 or 4 laboratory abnormalities (≥5%) in patients who received RETEVMO were decreased lymphocytes, increased ALT, decreased neutrophils, increased ALP, increased blood creatinine, decreased calcium, and increased AST.
Table 12 summarizes the adverse reactions in LIBRETTO-531.
|
1 Hypertension includes hypertension, blood pressure increased. |
||||
|
2 Edema includes edema peripheral, face edema, periorbital edema, swelling face, peripheral swelling, localized edema, eyelid edema, generalized edema, eye swelling, lymphoedema, orbital edema, eye edema, edema, edema genital, swelling, scrotal edema, scrotal swelling, angioedema, skin edema, testicular swelling, vulvovaginal swelling. |
||||
|
3 Fatigue includes fatigue, asthenia, malaise. |
||||
|
4 Dry mouth includes dry mouth, mucosal dryness. |
||||
|
5 Diarrhea includes diarrhea, anal incontinence, defecation urgency, frequent bowel movements, gastrointestinal hypermotility. |
||||
|
6 Abdominal pain included abdominal pain, abdominal pain upper, abdominal discomfort, abdominal pain lower, gastrointestinal pain. |
||||
|
7 Stomatitis includes stomatitis, mouth ulceration, mucosal inflammation. |
||||
|
8 Headache includes headache, sinus headache, tension headache. |
||||
|
9 Rash includes rash, rash maculopapular, skin exfoliation, rash erythematous, rash macular, dermatitis, urticaria, rash pruritic, exfoliative rash, rash papular, dermatitis allergic, rash follicular, rash generalized, rash pustular, butterfly rash, rash morbilliform, rash vesicular. |
||||
|
10 Electrocardiogram QT prolongation includes electrocardiogram QT prolonged, electrocardiogram QT interval abnormal. |
||||
|
11 Hypothyroidism includes hypothyroidism, blood thyroid stimulating hormone increased. |
||||
|
* Only includes a Grade 3 adverse reaction |
||||
|
# Graded according to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) Version 5.0. |
||||
| Adverse Reaction | RETEVMO N = 193 |
Cabozantinib or Vandetanib N = 97 |
||
| Grades 1-4# (%) |
Grades 3-4 (%) |
Grades 1-4# (%) |
Grades 3-4 (%) |
|
| Vascular disorders | ||||
| Hypertension1 | 43 | 19* | 41 | 18* |
| General disorders and administration-site conditions | ||||
| Edema2 | 33 | 0 | 5 | 0 |
| Fatigue3 | 28 | 4.1* | 47 | 9* |
| Pyrexia | 12 | 1.0* | 2.1 | 0 |
| Gastrointestinal disorders | ||||
| Dry mouth4 | 32 | 0.5* | 10 | 1.0* |
| Diarrhea5 | 26 | 3.1* | 61 | 8* |
| Abdominal pain6 | 18 | 0.5* | 21 | 2.1* |
| Constipation | 16 | 0 | 12 | 0 |
| Stomatitis7 | 14 | 0.5* | 42 | 13* |
| Nausea | 10 | 1.0* | 32 | 5* |
| Nervous system disorders | ||||
| Headache8 | 23 | 0.5* | 21 | 0 |
| Skin and subcutaneous tissue disorders | ||||
| Rash9 | 19 | 1.6* | 27 | 4.1* |
| Reproductive system and breast disorders | ||||
| Erectile dysfunction | 16 | 0 | 0 | 0 |
| Investigations | ||||
| Electrocardiogram QT prolonged10 | 14 | 4.7* | 13 | 2.1* |
| Metabolism and nutrition disorders | ||||
| Decreased appetite | 12 | 0.5* | 28 | 5* |
| Endocrine disorders | ||||
| Hypothyroidism11 | 11 | 0 | 21 | 0 |
Clinically relevant adverse reactions in ≤10% of patients who received RETEVMO include dizziness (8%); urinary tract infections (8%); vomiting (8%); pneumonia, interstitial lung disease/pneumonitis, chylous ascites and hypersensitivity (all < 2%).
Table 13 summarizes the laboratory abnormalities in LIBRETTO-531.
|
1 Denominator for each laboratory parameter is based on the number of patients with a baseline and post-treatment laboratory value available: RETEVMO (range: 183 to 191 patients) and chemotherapy with or without cabozantinib or vandetanib (range: 91 to 94 patients). |
||||
|
* Only includes a Grade 3 laboratory abnormality |
||||
|
# Graded according to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 5.0 |
||||
| Laboratory Abnormality | RETEVMO1 | Cabozantinib or Vandetanib1 | ||
| Grades 1-4# % |
Grades 3-4 % |
Grades 1-4# % |
Grades 3-4 % |
|
| Chemistry | ||||
| Calcium decreased | 55 | 5 | 62 | 11 |
| ALT increased | 53 | 16 | 72 | 7* |
| AST increased | 47 | 5 | 68 | 3.2* |
| Alkaline phosphatase increased | 37 | 6 | 28 | 5 |
| Total bilirubin increased | 32 | 1.1 | 30 | 3.2* |
| Blood creatinine increased | 27 | 6 | 16 | 8 |
| Sodium decreased | 20 | 3.2* | 16 | 0 |
| Albumin decreased | 11 | 1.1 | 7 | 0 |
| Magnesium decreased | 9 | 3.3 | 26 | 9 |
| Potassium decreased | 8 | 0 | 22 | 4.4* |
| Hematology | ||||
| Lymphocyte count decreased | 41 | 18 | 36 | 13 |
| Neutrophil count decreased | 33 | 14 | 42 | 19 |
| Platelets decreased | 28 | 1.1 | 34 | 1.1* |
| Hemoglobin decreased | 18 | 2.1* | 23 | 2.1* |
Increased Creatinine
In healthy subjects administered RETEVMO 160 mg orally twice daily, serum creatinine increased 18% after 10 days. Consider alternative markers of renal function if persistent elevations in serum creatinine are observed [see Clinical Pharmacology (12.3)].
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on RETEVMO
Acid-Reducing Agents
Concomitant use of RETEVMO with acid-reducing agents decreases selpercatinib plasma concentrations [see Clinical Pharmacology (12.3)], which may reduce RETEVMO anti-tumor activity.
Avoid concomitant use of PPIs, H2 receptor antagonists, and locally-acting antacids with RETEVMO. If coadministration cannot be avoided, take RETEVMO with food (with a PPI) or modify its administration time (with a H2 receptor antagonist or a locally-acting antacid) [see Dosage and Administration (2.4)].
Strong and Moderate CYP3A Inhibitors
Concomitant use of RETEVMO with a strong or moderate CYP3A inhibitor increases selpercatinib plasma concentrations [see Clinical Pharmacology (12.3)], which may increase the risk of RETEVMO adverse reactions, including QTc interval prolongation.
Avoid concomitant use of strong and moderate CYP3A inhibitors with RETEVMO. If concomitant use of strong and moderate CYP3A inhibitors cannot be avoided, reduce the RETEVMO dosage and monitor the QT interval with ECGs more frequently [see Dosage and Administration (2.6), Warning and Precautions (5.4)].
Strong and Moderate CYP3A Inducers
Concomitant use of RETEVMO with a strong or moderate CYP3A inducer decreases selpercatinib plasma concentrations [see Clinical Pharmacology (12.3)], which may reduce RETEVMO anti-tumor activity.
Avoid coadministration of strong or moderate CYP3A inducers with RETEVMO.
7.2 Effects of RETEVMO on Other Drugs
CYP2C8 and CYP3A Substrates
RETEVMO is a moderate CYP2C8 inhibitor and a weak CYP3A inhibitor. Concomitant use of RETEVMO with CYP2C8 and CYP3A substrates increases their plasma concentrations [see Clinical Pharmacology (12.3)], which may increase the risk of adverse reactions related to these substrates. Avoid coadministration of RETEVMO with CYP2C8 and CYP3A substrates where minimal concentration changes may lead to increased adverse reactions. If coadministration cannot be avoided, follow recommendations for CYP2C8 and CYP3A substrates provided in their approved product labeling.
Certain P-gp Substrates
RETEVMO is a P-gp inhibitor. Concomitant use of RETEVMO with P-gp substrates increases their plasma concentrations [see Clinical Pharmacology (12.3)], which may increase the risk of adverse reactions related to these substrates. Avoid coadministration of RETEVMO with P-gp substrates where minimal concentration changes may lead to increased adverse reactions. If coadministration cannot be avoided, follow recommendations for P-gp substrates provided in their approved product labeling.
7.3 Drugs that Prolong QT Interval
RETEVMO is associated with QTc interval prolongation [see Warnings and Precautions (5.4), Clinical Pharmacology (12.2)]. Monitor the QT interval with ECGs more frequently in patients who require treatment with concomitant medications known to prolong the QT interval.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies, and its mechanism of action [see Clinical Pharmacology (12.1)], RETEVMO can cause fetal harm when administered to a pregnant woman. There are no available data on RETEVMO use in pregnant women to inform drug-associated risk. Administration of selpercatinib to pregnant rats during the period of organogenesis resulted in embryolethality and malformations at maternal exposures that were approximately equal to the human exposure at the clinical dose of 160 mg twice daily. Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
Selpercatinib administration to pregnant rats during the period of organogenesis at oral doses ≥100 mg/kg [approximately 3.6 times the human exposure based on the area under the curve (AUC) at the clinical dose of 160 mg twice daily] resulted in 100% post-implantation loss. At the dose of 50 mg/kg [approximately equal to the human exposure (AUC) at the clinical dose of 160 mg twice daily], 6 of 8 females had 100% early resorptions; the remaining 2 females had high levels of early resorptions with only 3 viable fetuses across the 2 litters. All viable fetuses had decreased fetal body weight and malformations (2 with short tail and one with small snout and localized edema of the neck and thorax).
8.2 Lactation
Risk Summary
There are no data on the presence of selpercatinib or its metabolites in human milk or on their effects on the breastfed child or on milk production. Because of the potential for serious adverse reactions in breastfed children, advise women not to breastfeed during treatment with RETEVMO and for 1 week after the last dose.
8.3 Females and Males of Reproductive Potential
Based on animal data, RETEVMO can cause embryolethality and malformations at doses resulting in exposures less than or equal to the human exposure at the clinical dose of 160 mg twice daily [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to initiating RETEVMO [see Use in Specific Populations (8.1)].
8.4 Pediatric Use
The safety and effectiveness of RETEVMO have been established in pediatric patients 2 years of age and older for the treatment of:
- advanced or metastatic medullary thyroid cancer (MTC) with a RET mutation who require systemic therapy
- advanced or metastatic thyroid cancer with a RET gene fusion who require systemic therapy and are radioactive iodine-refractory (if radioactive iodine is appropriate)
- locally advanced or metastatic solid tumors with a RET gene fusion that have progressed on or following prior systemic treatment or who have no satisfactory alternative treatment options.
Use of RETEVMO for these indications is supported by evidence from adequate and well-controlled studies in adult and pediatric patients with additional pharmacokinetic and safety data in pediatric patients 2 years of age and older [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), Clinical Studies (14.2, 14.3, 14.4)]. The predicted exposures of selpercatinib in pediatric patients at the recommended dosages were within the range of values predicted in patients ≥ 12 years and ≥ 50 kg in body weight receiving the approved recommended dosage of 160 mg twice daily [see Clinical Pharmacology (12.3)].
The safety and effectiveness of RETEVMO have not been established in these indications in patients less than 2 years of age.
The safety and effectiveness of RETEVMO have not been established in pediatric patients for other indications [see Indications and Usage (1)].
Juvenile Animal Toxicity Data
In a juvenile rat toxicity study, animals were dosed daily with selpercatinib from post-natal day 21 to day 70 (approximately equivalent to a human child to late adolescent). Selpercatinib increased physeal thickness of multiple bones, extending into the metaphysis and associated with decreased trabecular bone, which was not reversible at doses approximately equivalent to or greater than the adult human exposure at the clinical dose of 160 mg twice daily. Growth plate changes were associated with impairment of bone modeling, resulting in decreased femur length and with reduction in bone mineral density. Selpercatinib also induced reversible hypocellularity of bone marrow in males at ≥30 mg/kg (approximately equivalent to or greater than the adult human exposure at the clinical dose of 160 mg twice daily), and reversible alterations of dentin composition at ≥50 mg/kg (approximately 3 times the adult human exposure at the clinical dose of 160 mg twice daily). Irreversible, dose-dependent degeneration of testicular germinal epithelium, with vacuolation of Sertoli cells and corresponding depletion of spermatozoa in the epididymides, was also observed at ≥ 30 mg/kg (approximately equivalent to or greater than the adult human exposure at the clinical dose of 160 mg twice daily) and affected male reproductive performance at 50 mg/kg (approximately 3 times the adult human exposure at the clinical dose of 160 mg twice daily). Females exhibited delay in attainment of vaginal patency, a marker of sexual maturity, at 125 mg/kg (approximately 4 times the adult human exposure at the clinical dose of 160 mg twice daily); this effect was associated with lower mean body weight. Similar effects in irregular thickening of growth plates in adult rats and minipigs, and tooth dysplasia and malocclusion, resulting in tooth loss in adult rats were observed in repeat dose studies of up to 13-week duration with selpercatinib.
Monitor growth plates in pediatric patients with open growth plates. Consider interrupting or discontinuing therapy based on the severity of any growth plate abnormalities and based on an individual risk-benefit assessment.
8.5 Geriatric Use
Of 796 patients who received RETEVMO, 34% (268 patients) were ≥65 years of age and 9% (74 patients) were ≥75 years of age. No overall differences were observed in the safety or effectiveness of RETEVMO between patients who were ≥65 years of age and younger patients.
8.6 Renal Impairment
No dosage modification is recommended for patients with mild to severe renal impairment [estimated Glomerular Filtration Rate (eGFR) ≥15 to 89 mL/min, estimated by Modification of Diet in Renal Disease (MDRD) equation]. The recommended dosage has not been established for patients with end-stage renal disease (ESRD) [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
Reduce the dose when administering RETEVMO to patients with severe [total bilirubin greater than 3 to 10 times upper limit of normal (ULN) and any AST] hepatic impairment [see Dosage and Administration (2.7)]. No dosage modification is recommended for patients with mild (total bilirubin less than or equal to ULN with AST greater than ULN or total bilirubin greater than 1 to 1.5 times ULN with any AST) or moderate (total bilirubin greater than 1.5 to 3 times ULN and any AST) hepatic impairment. Monitor for RETEVMO-related adverse reactions in patients with hepatic impairment [see Clinical Pharmacology (12.3)].
11 DESCRIPTION
RETEVMO contains selpercatinib, a kinase inhibitor. The molecular formula for selpercatinib is C29H31N7O3 and the molecular weight is 525.61 g/mol. The chemical name is 6-(2-hydroxy-2-methylpropoxy)-4-(6-(6-((6-methoxypyridin-3-yl)methyl)-3,6-diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)pyrazolo[1,5-a]pyridine-3-carbonitrile. Selpercatinib has the following chemical structure:
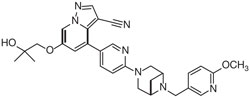
Selpercatinib is a white to light yellow powder that is slightly hygroscopic. The aqueous solubility of selpercatinib is pH dependent, from sparingly soluble at low pH to practically insoluble at neutral pH.
RETEVMO capsules contain either 40 mg or 80 mg of selpercatinib in hard gelatin capsules for oral use. Each capsule contains inactive ingredients of colloidal silicon dioxide and microcrystalline cellulose. The 40 mg capsule shell is composed of gelatin, titanium dioxide, ferric oxide black and black ink. The 80 mg capsule shell is composed of gelatin, titanium dioxide, FD&C blue #1 and black ink. The black ink is composed of shellac, potassium hydroxide and ferric oxide black.
RETEVMO tablets contain 40 mg, 80 mg, 120 mg or 160 mg of selpercatinib as film coated, debossed tablets for oral use. Each tablet contains inactive ingredients of croscarmellose sodium, hydroxypropyl cellulose, mannitol, microcrystalline cellulose, and sodium stearyl fumarate. The tablet film coating material contains polyvinyl alcohol, titanium dioxide, polyethylene glycol, and talc. Additionally, the film coating of the 40 mg, 80 mg, and 120 mg tablets contains ferrosoferric oxide and the film coating of the 80 mg, 120 mg, and 160 mg tablets contain ferric oxide.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Selpercatinib is a kinase inhibitor. Selpercatinib inhibited wild-type RET and multiple mutated RET isoforms as well as VEGFR1 and VEGFR3 with IC50 values ranging from 0.92 nM to 67.8 nM. In other enzyme assays, selpercatinib also inhibited FGFR 1, 2, and 3 at higher concentrations that were still clinically achievable. In cellular assays, selpercatinib inhibited RET at approximately 60-fold lower concentrations than FGFR1 and 2 and approximately 8-fold lower concentration than VEGFR3.
Certain point mutations in RET or chromosomal rearrangements involving in-frame fusions of RET with various partners can result in constitutively activated chimeric RET fusion proteins that can act as oncogenic drivers by promoting cell proliferation of tumor cell lines. In in vitro and in vivo tumor models, selpercatinib demonstrated anti-tumor activity in cells harboring constitutive activation of RET proteins resulting from gene fusions and mutations, including CCDC6-RET, KIF5B-RET, RET V804M, and RET M918T. In addition, selpercatinib showed anti-tumor activity in mice intracranially implanted with a patient-derived RET fusion positive tumor.
12.2 Pharmacodynamics
Exposure-Response Relationship
Selpercatinib exposure-response relationships and the time course of pharmacodynamic response have not been fully characterized.
Cardiac Electrophysiology
The effect of RETEVMO on the QTc interval was evaluated in a thorough QT study in healthy subjects. The largest mean increase in QTc is predicted to be 10.6 msec (upper 90% confidence interval: 12.1 msec) at the mean steady-state maximum concentration (Cmax) observed in patients after administration of 160 mg twice daily. The increase in QTc was concentration-dependent.
12.3 Pharmacokinetics
The pharmacokinetics of selpercatinib capsules were evaluated in patients with locally advanced or metastatic solid tumors administered 160 mg twice daily unless otherwise specified. The capsule and tablet dosage forms of selpercatinib are bioequivalent. Steady state selpercatinib AUC and Cmax increased in a slightly greater than dose proportional manner over the dose range of 20 mg once daily to 240 mg twice daily [0.06 to 1.5 times the maximum recommended total daily dosage].
Steady-state was reached by approximately 7 days and the median accumulation ratio after administration of 160 mg twice daily was 3.4-fold. Mean steady-state selpercatinib [coefficient of variation (CV%)] Cmax was 2,980 (53%) ng/mL and AUC0-24h was 51,600 (58%) ng*h/mL.
Absorption
The median tmax of selpercatinib is 2 hours. The mean absolute bioavailability of RETEVMO capsules is 73% (60% to 82%) in healthy subjects.
Distribution
The apparent volume of distribution (Vss/F) of selpercatinib is 203 L.
Protein binding of selpercatinib is 96% in vitro and is independent of concentration. The blood-to-plasma concentration ratio is 0.7.
Elimination
The apparent clearance (CL/F) of selpercatinib is 6 L/h in patients and the half-life is 32 hours following oral administration of RETEVMO in healthy subjects.
Specific Populations
The apparent volume of distribution and clearance of selpercatinib increase with increasing body weight (9.6 kg to 179 kg).
No clinically significant differences in the pharmacokinetics of selpercatinib were observed based on age (2 years to 92 years), sex, or mild, moderate, or severe renal impairment (eGFR ≥15 to 89 mL/min). The effect of ESRD on selpercatinib pharmacokinetics has not been studied.
Pediatric patients
The exposures of selpercatinib in pediatric patients are predicted to be comparable to those in adult patients administered at the recommended dosages.
Patients with Hepatic Impairment
The selpercatinib AUC0-INF increased 1.07-fold, 1.32-fold, and 1.77-fold in subjects with mild (total bilirubin less than or equal to ULN with AST greater than ULN or total bilirubin greater than 1 to 1.5 times ULN with any AST), moderate (total bilirubin greater than 1.5 to 3 times ULN and any AST), and severe (total bilirubin greater than 3 to 10 times ULN and any AST) hepatic impairment, respectively, compared to subjects with normal hepatic function.
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
Proton-Pump Inhibitors (PPI): Coadministration with multiple daily doses of omeprazole (PPI) decreased selpercatinib AUC0-INF and Cmax when RETEVMO was administered fasting. Coadministration with multiple daily doses of omeprazole did not significantly change the selpercatinib AUC0-INF and Cmax when RETEVMO was administered with food (Table 14).
|
1 High-fat meal: approximately 150, 250, and 500-600 calories from protein, carbohydrate, and fat, respectively; approximately 800 to 1,000 calories total. |
||
|
2 Low-fat meal: approximately 390 calories and 10 g of fat. |
||
| Selpercatinib AUC0-INF |
Selpercatinib Cmax |
|
| RETEVMO fasting | Reference | Reference |
| RETEVMO fasting + PPI | ↓ 69% | ↓ 88% |
| RETEVMO with a high-fat meal1 + PPI | ↑ 2% | ↓ 49% |
| RETEVMO with a low-fat meal2 + PPI | No change | ↓ 22% |
H2 Receptor Antagonists: No clinically significant differences in selpercatinib pharmacokinetics were observed when coadministered with multiple daily doses of ranitidine (H2 receptor antagonist) given 10 hours prior to and 2 hours after the RETEVMO dose (administered fasting).
Strong CYP3A Inhibitors: Coadministration of multiple doses of itraconazole (strong CYP3A inhibitor) increased the selpercatinib AUC0-INF 2.33-fold and Cmax 1.3-fold.
Moderate CYP3A Inhibitors: Coadministration of multiple doses of diltiazem, fluconazole, or verapamil (moderate CYP3A inhibitors) is predicted to increase the selpercatinib AUC 1.6 to 1.99-fold and Cmax 1.46 to 1.76-fold.
Strong CYP3A Inducers: Coadministration of multiple doses of rifampin (strong CYP3A inducer) decreased the selpercatinib AUC0-INF by 87% and Cmax by 70%.
Moderate CYP3A Inducers: Coadministration of multiple doses of bosentan or efavirenz (moderate CYP3A inducers) is predicted to decrease the selpercatinib AUC by 40-70% and Cmax by 34-57%.
Weak CYP3A Inducers: Coadministration of multiple doses of modafinil (weak CYP3A inducer) is predicted to decrease the selpercatinib AUC by 33% and Cmax by 26%.
CYP2C8 Substrates: Coadministration of RETEVMO with repaglinide (sensitive CYP2C8 substrate) increased the repaglinide AUC0-INF 2.88-fold and Cmax 1.91-fold.
CYP3A Substrates: Coadministration of RETEVMO with midazolam (sensitive CYP3A substrate) increased the midazolam AUC0-INF 1.54-fold and Cmax 1.39-fold.
P-glycoprotein (P-gp) Substrates: Coadministration of RETEVMO with dabigatran (P-gp substrate) increased the dabigatran AUC0-INF 1.38-fold and Cmax 1.43-fold.
P-gp Inhibitors: No clinically significant differences in selpercatinib pharmacokinetics were observed when coadministered with a single dose of rifampin (P-gp inhibitor).
MATE1 Substrates: No clinically significant differences in glucose levels were observed when metformin (MATE1 substrate) was coadministered with selpercatinib.
In Vitro Studies
CYP Enzymes: Selpercatinib does not inhibit or induce CYP1A2, CYP2B6, CYP2C9, CYP2C19, or CYP2D6 at clinically relevant concentrations.
Transporter Systems: Selpercatinib inhibits MATE1 and BCRP, but does not inhibit OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, BSEP, and MATE2-K at clinically relevant concentrations. Selpercatinib may increase serum creatinine by decreasing renal tubular secretion of creatinine via inhibition of MATE1 [see Adverse Effects (6.1)]. Selpercatinib is a substrate for P-gp and BCRP, but not for OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, MATE1, or MATE2-K.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Selpercatinib was not carcinogenic in a 2-year study in rats when administered by daily oral gavage at doses up to 20 mg/kg in males or 40 mg/kg in females (approximately equal to the human exposure by AUC at the 160 mg twice daily clinical dose). Selpercatinib was not carcinogenic in a 6-month study in rasH2 transgenic mice when administered by daily oral gavage at doses of up to 60 mg/kg.
Selpercatinib was not mutagenic in the in vitro bacterial reverse mutation (Ames) assays, with or without metabolic activation, or clastogenic in the in vitro micronucleus assay in human peripheral lymphocytes, with or without metabolic activation. Selpercatinib was positive in the in vivo micronucleus assay in rats at concentrations >7 times the Cmax at the human dose of 160 mg twice daily.
In general toxicology studies, male rats and minipigs exhibited testicular degeneration which was associated with luminal cell debris and/or reduced luminal sperm in the epididymis at selpercatinib exposures approximately 0.4 (rat) and 0.1 (minipig) times the clinical exposure by AUC at the 160 twice daily clinical dose. In a dedicated fertility study in male rats, administration of selpercatinib at doses up to 30 mg/kg/day (approximately twice the clinical exposure by AUC at the 160 twice daily clinical dose) for 28 days prior to cohabitation with untreated females did not affect mating or have clear effects on fertility. Males did, however, display a dose-dependent increase in testicular germ cell depletion and spermatid retention at doses ≥3 mg/kg (~0.2 times the clinical exposure by AUC at the 160 twice daily clinical dose) accompanied by altered sperm morphology at 30 mg/kg.
In a dedicated fertility study in female rats treated with selpercatinib for 15 days before mating to Gestational Day 7, there were decreases in the number of estrous cycles at a dose of 75 mg/kg (approximately equal to the human exposure by AUC at the 160 mg twice daily clinical dose). While selpercatinib did not have clear effects on mating performance or ability to become pregnant at any dose level, half of females at the 75 mg/kg dose level had 100% nonviable embryos. At the same dose level in females with some viable embryos there were increases in post-implantation loss. In a 3-month general toxicology study in minipigs, there were findings of decreased or absent corpora lutea at a selpercatinib dose of 15 mg/kg (approximately 0.3 times to the human exposure by AUC at the 160 mg twice daily clinical dose). Corpora luteal cysts were present in the minipig at selpercatinib doses ≥2 mg/kg (approximately 0.07 times the human exposure by AUC at the 160 mg twice daily clinical dose).
14 CLINICAL STUDIES
14.1 RET Fusion-Positive Non-Small Cell Lung Cancer
LIBRETTO-001
The efficacy of RETEVMO was evaluated in patients with advanced RET fusion-positive NSCLC enrolled in a multicenter, open-label, multi-cohort clinical trial (LIBRETTO-001, NCT03157128). The study enrolled patients with advanced or metastatic RET fusion-positive NSCLC who had progressed on platinum-based chemotherapy and patients with locally advanced (stage III who were not candidates for surgical resection or definitive chemoradiation) or metastatic NSCLC without prior systemic therapy in separate cohorts. Identification of a RET gene alteration was prospectively determined in local laboratories using next generation sequencing (NGS), polymerase chain reaction (PCR), fluorescence in situ hybridization (FISH) or other local testing methods. Adult patients received RETEVMO 160 mg orally twice daily until unacceptable toxicity or disease progression; patients enrolled in the dose escalation phase were permitted to adjust their dose to 160 mg twice daily. The major efficacy outcome measures were confirmed overall response rate (ORR) and duration of response (DOR), as determined by a blinded independent review committee (BIRC) according to RECIST v1.1.
RET Fusion-Positive NSCLC Previously Treated with Platinum Chemotherapy
Efficacy was evaluated in 247 patients with RET fusion-positive NSCLC previously treated with platinum chemotherapy enrolled into a cohort of LIBRETTO-001.
The median age was 61 years (range: 23 to 81); 57% were female; 44% were White, 48% were Asian, 4.9% were Black or African American; and 2.8% were Hispanic/Latino. ECOG performance status was 0-1 (97%) or 2 (3%) and 97% of patients had metastatic disease. Patients received a median of 2 prior systemic therapies (range 1–15); 58% had prior anti-PD1/PD-L1 therapy. RET fusions were detected in 94% of patients using NGS (84.6% tumor samples; 9.3% blood or plasma samples), 4.0% using FISH, 1.6% using PCR and 0.4% by other local testing methods.
Efficacy results for previously treated RET fusion-positive NSCLC are summarized in Table 15.
|
1 Confirmed overall response rate assessed by BIRC. |
|
|
2 Based on observed duration of response. |
|
|
NE = not estimable |
|
| RETEVMO (n = 247) |
|
| Overall Response Rate1(95% CI) | 61% (55%, 67%) |
| Complete response | 7.3% |
| Partial response | 54% |
| Duration of Response | |
| Median in months (95% CI) | 28.6 (20, NE) |
| % with ≥ 12 months2 | 63% |
For the 144 patients who received an anti-PD-1 or anti-PD-L1 therapy, either sequentially or concurrently with platinum-based chemotherapy, an exploratory subgroup analysis of ORR was 63% (95% CI: 54%, 70%) and the median DOR was 28.6 months (95% CI: 14.8, NE).
Among the 247 patients with previously treated RET fusion-positive NSCLC, 16 had measurable CNS metastases at baseline as assessed by BIRC. One patient received radiation therapy (RT) to the brain within 2 months prior to study entry. Responses in intracranial lesions were observed in 14 of these 16 patients; 39% of responders had an intracranial DOR of ≥ 12 months.
Treatment-naïveRETFusion-Positive NSCLC
Efficacy was evaluated in 69 patients with treatment-naïve RET fusion-positive NSCLC enrolled into a cohort of LIBRETTO-001.
The median age was 63 years (range 23 to 92); 62% were female; 70% were White, 19% were Asian, and 6% were Black or African American. ECOG performance status was 0-1 (94%) or 2 (6%) and 99% of patients had metastatic disease. RET fusions were detected in 91% of patients using NGS (60.9% tumor samples; 30.4% in blood), 7.2% using FISH and 1.4% using PCR.
Efficacy results for treatment naïve RET fusion-positive NSCLC are summarized in Table 16.
|
1 Confirmed overall response rate assessed by BIRC. |
|
|
2 Based on observed duration of response. |
|
|
NE = not estimable |
|
| RETEVMO (n =69) |
|
| Overall Response Rate1(95% CI) | 84% (73%, 92%) |
| Complete response | 5.8% |
| Partial response | 78% |
| Duration of Response | |
| Median in months (95% CI) | 20.2 (13, NE) |
| % with ≥ 12 months2 | 50% |
Among the 69 patients with treatment-naïve RET fusion-positive NSCLC, 5 had measurable CNS metastases at baseline as assessed by BIRC. Two patients received RT to the brain within 2 months prior to study entry. Responses in intracranial lesions were observed in 4 of these 5 patients; 38% of responders had an intracranial DOR of ≥ 12 months.
LIBRETTO-431
The efficacy of RETEVMO was evaluated in patients with unresectable, locally advanced or metastatic, RET fusion-positive NSCLC enrolled in a multicenter, open-label, active-controlled, randomized trial (LIBRETTO-431, NCT04194944). The trial evaluated RETEVMO compared to platinum-based and pemetrexed chemotherapy with or without pembrolizumab in patients with RET fusion-positive, unresectable locally advanced or metastatic NSCLC with no previous systemic therapy for metastatic disease.
Patients (N=261) were randomized to receive either RETEVMO (160 mg orally twice daily) in continuous 21-day cycles or pemetrexed intravenously (IV) (500 mg per square meter of body-surface area) along with the investigator’s choice of platinum therapy (carboplatin IV [AUC 5, maximum dose 750 mg] or cisplatin IV [75 mg per square meter]) with or without pembrolizumab IV (200 mg) every 21 days. Treatment continued until disease progression or unacceptable toxicity. Crossover from the control arm to RETEVMO was permitted following disease progression. Patients were stratified according to geographic region (East Asia vs. elsewhere), brain metastases at baseline (presence vs. absence or unknown), and the investigator’s intent (before randomization) to treat the patient with or without pembrolizumab. Tumor assessments were performed every 6 weeks for two assessments, then every 9 weeks for four assessments, and then every 12 weeks thereafter.
The major efficacy outcome measure was progression-free survival (PFS) in patients intended to be treated with chemotherapy in combination with pembrolizumab and in the overall study population as determined by a blinded independent review committee (BIRC) according to RECIST v1.1. Other efficacy outcome measures included overall survival (OS) and overall response rate (ORR).
A total of 212 patients were enrolled in LIBRETTO-431 with an intent to treat with pembrolizumab if randomized to the control arm (129 into RETEVMO arm and 83 into chemotherapy with pembrolizumab arm). The median age was 61.5 years (range: 31 to 84 years); 47% were male; 41% White, 55% Asian, and 0.9% Black or African American, 1.4% American Indian or Alaska Native, 1.9% were race not reported; ethnicity was not reported in 96% of patients. ECOG performance status was 0-1 (97%) or 2 (3%), 68% were never smokers, 93% of patients had metastatic disease, and 14% had measurable intracranial metastases at baseline, as determined by a neuroradiologic BIRC. RET fusions were detected in 60% of patients using NGS and 40% using PCR (89% tumor samples; 11% in blood).
Efficacy results from the pre-planned interim efficacy analysis are summarized in Table 17.
|
1 Based on the stratified Cox proportional hazard model, stratified by geographic location (East Asia versus elsewhere), brain metastases at baseline according to investigator (presence versus absence or unknown). |
||
|
2 Based on stratified log-rank test, stratified by geographic location (East Asia versus elsewhere), brain metastases at baseline according to investigator (presence versus absence or unknown). |
||
|
3 Based on observed duration of response. |
||
|
NE = not estimable |
||
| RETEVMO (n = 129) |
Chemotherapy with pembrolizumab (n = 83) |
|
| Progression-Free Survival | ||
| Number (%) of patients with an event | 49 (38%) | 49 (59%) |
| Medians in months (95% CI) | 24.8 (16.9, NE) | 11.2 (8.8, 16.8) |
| Hazard ratio1 (95% CI) | 0.46 (0.31, 0.70) | |
| p-value2 | 0.0002 | |
| Overall Response Rate (95% CI) | 84% (76, 90) | 65% (54, 75) |
| Complete response | 7% | 6% |
| Partial response | 77% | 59% |
| Duration of Response | ||
| Median in months (95% CI) % with ≥ 12 months3 |
24.2 (17.9, NE) 60% |
11.5 (9.7, 23.3) 30% |
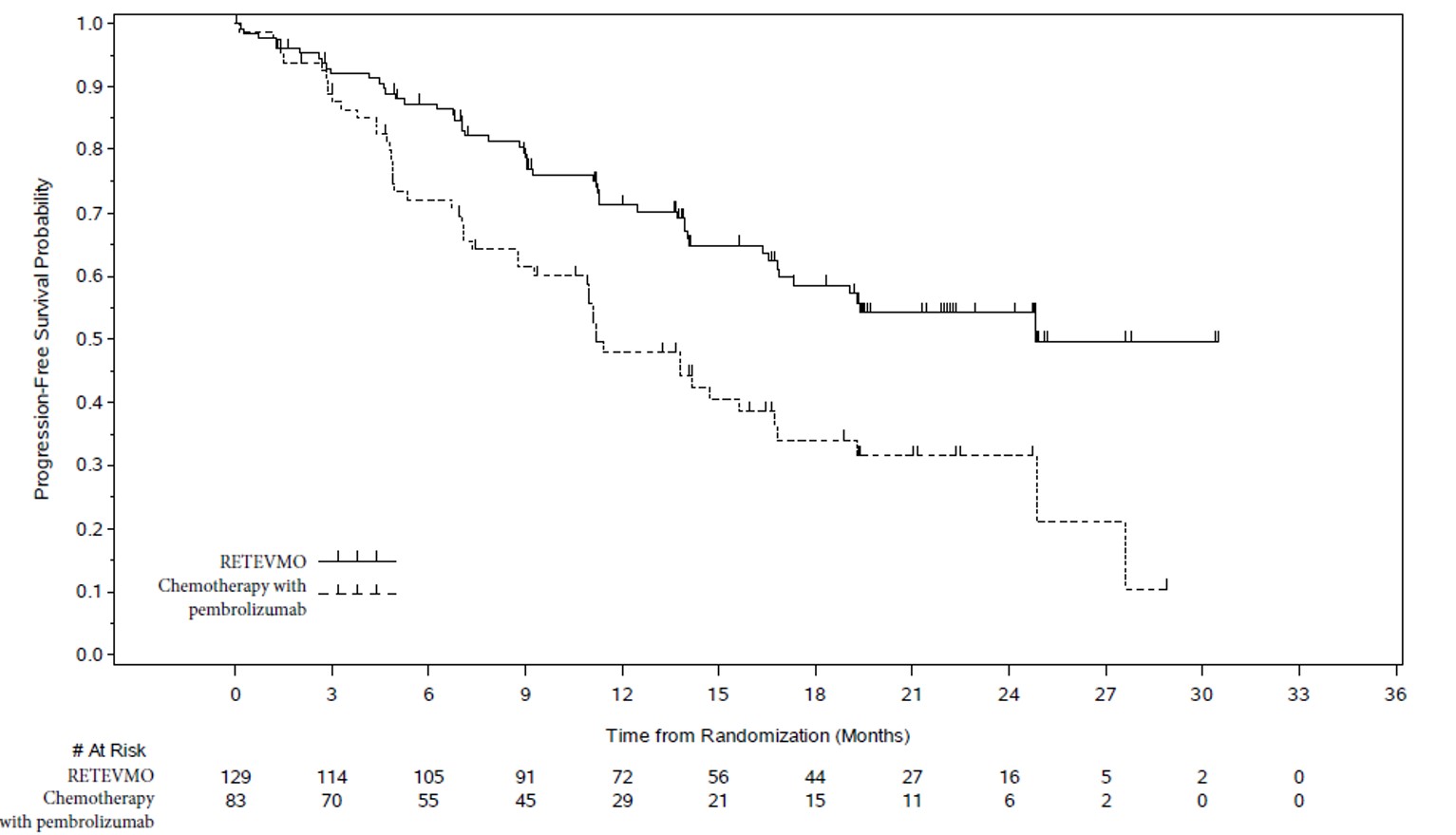
Figure 1: Kaplan-Meier Curves of Progression-Free Survival in LIBRETTO-431: RETEVMO versus Chemotherapy with Pembrolizumab
Among the 212 randomized patients, 29 had measurable CNS metastases at baseline as assessed by BIRC. Responses in intracranial lesions were observed in 14 of 17 patients treated with RETEVMO and 7 of 12 patients treated with chemotherapy with pembrolizumab.
Overall survival was immature at the time of the PFS interim analysis. At the time of an updated descriptive analysis of OS (43% of prespecified OS events needed for the final analysis), a total of 49 (31%) and 26 (25%) patients died in the RETEVMO and the control arm, respectively. The OS HR was 1.26 (95% CI: 0.78, 2.04). Overall survival may be affected by the imbalance in post-progression therapies. Of 68 control arm patients who had disease progression, 50 patients (74%) received RETEVMO at progression. Of 71 RETEVMO arm patients who had disease progression, 16 (23%) received chemotherapy and/or immune checkpoint inhibitor therapy, and 44 (62%) continued receiving RETEVMO.
14.2 RET-Mutant Medullary Thyroid Cancer
LIBRETTO-001
The efficacy of RETEVMO was evaluated in patients with RET-mutant MTC enrolled in a multicenter, open-label, multi-cohort clinical trial (NCT03157128). The study enrolled patients with advanced or metastatic RET-mutant MTC who had been previously treated with cabozantinib or vandetanib (or both) and patients with advanced or metastatic RET-mutant MTC who were naïve to cabozantinib and vandetanib in separate cohorts.
RET-Mutant MTC Previously Treated with Cabozantinib or Vandetanib
Efficacy was evaluated in 55 patients with RET-mutant advanced MTC who had previously treated with cabozantinib or vandetanib enrolled into a cohort of LIBRETTO-001.
The median age was 57 years (range: 17 to 84); 66% were male; 89% were White, 7% were Hispanic/Latino, and 1.8% were Black. ECOG performance status was 0-1 (95%) or 2 (5%) and 98% of patients had metastatic disease. Patients received a median of 2 prior systemic therapies (range 1 – 8). RET mutation status was detected in 82% of patients using NGS (78% tumor samples; 4% blood or plasma), 16% using PCR, and 2% using an unknown test. The protocol excluded patients with synonymous, frameshift or nonsense RET mutations; the specific mutations used to identify and enroll patients are described in Table 18.
|
1 Somatic or germline mutations; protein change. |
|||
|
2 Extracellular cysteine mutations involving cysteine residues 609, 611, 618, 620, 630, and 634. |
|||
|
3 Other included: K666N (1), D631_L633delinsV (2), D631_L633delinsE (5), D378_G385delinsE (1), D898_E901del (2), A883F (4), E632_L633del (4), L790F (2), T636_V637insCRT(1), D898_E901del + D903_S904delinsEP (1). |
|||
|
4 One patient also had a M918T mutation. |
|||
| RET Mutation Type1 | Previously Treated (n = 55) |
Cabozantinib/ Vandetanib Naïve (n = 88) |
Total (n = 143) |
| M918T | 33 | 49 | 82 |
| Extracellular cysteine mutation2 | 7 | 20 | 27 |
| V804M or V804L | 54 | 6 | 11 |
| Other3 | 10 | 13 | 23 |
Efficacy results for RET-mutant MTC are summarized in Table 19.
|
1 Confirmed overall response rate assessed by BIRC. |
|
|
2 Based on observed duration of response. |
|
|
NE = not estimable |
|
| RETEVMO (n = 55) |
|
| Overall Response Rate1(95% CI) | 76% (63%, 87%) |
| Complete response | 18% |
| Partial response | 58% |
| Duration of Response | |
| Median in months (95% CI) | 45.3 (29.9, NE) |
| % with ≥12 months2 | 76% |
Cabozantinib and Vandetanib-naïveRET-Mutant MTC
Efficacy was evaluated in 88 patients with RET-mutant MTC who were cabozantinib and vandetanib treatment-naïve enrolled into a cohort of LIBRETTO-001.
The median age was 58 years (range: 15 to 82) with two patients (2.3%) aged 12 to 16 years; 66% were male; and 86% were White, 4.5% were Asian, and 2.3% were Hispanic/Latino. ECOG performance status was 0-1 (97%) or 2 (3.4%). All patients (100%) had metastatic disease and 18% had received 1 or 2 prior systemic therapies (including 8% kinase inhibitors, 4.5% chemotherapy, 2.3% anti-PD1/PD-L1 therapy, and 1.1% radioactive iodine). RET mutation status was detected in 77.3% of patients using NGS (75.0% tumor samples; 2.3% blood samples), 18.2% using PCR, and 4.5% using an unknown test. The mutations used to identify and enroll patients are described in Table 18.
Efficacy results for cabozantinib and vandetanib-naïve RET-mutant MTC are summarized in Table 20.
|
1 Confirmed overall response rate assessed by BIRC. |
|
|
2 Based on observed duration of response. |
|
|
NR = not reached, NE = not estimable |
|
| RETEVMO (n = 88) |
|
| Overall Response Rate1(95% CI) | 81% (71%, 88%) |
| Complete response | 28% |
| Partial response | 52% |
| Duration of Response | |
| Median in months (95% CI) | NR (51.3, NE) |
| % with ≥12 months2 | 90% |
LIBRETTO-531
LIBRETTO-531 was a randomized (2:1), multicenter, open-label study (NCT04211337) in adults and adolescents with advance or metastatic RET-mutant MTC. The study evaluated the efficacy of RETEVMO versus physicians’ choice of cabozantinib or vandetanib in patients with progressive, advanced, kinase inhibitor naïve, RET-mutant medullary thyroid cancer.
Patients were randomized to receive either RETEVMO (160 mg twice daily) or physicians’ choice of cabozantinib (140 mg once daily) or vandetanib (300 mg once daily). Patients were stratified based on RET mutation (M918T vs. other) and intended treatment if randomized to the control arm (cabozantinib vs. vandetanib). The primary outcome was progression-free survival (PFS), as determined by a blinded independent review committee (BIRC) according to RECIST v1.1.
The median age was 55 years (range: 12 to 84), 63% were male, 58% were White, 23% were Asian, 2.4% were Black or African American, and 17% had unknown race. ECOG performance status was 0-1 (98%) or 2 (1.0%) with 0.7% unknown status. 77% of patients had metastatic disease and 6 patients (2.1%) had received 1 prior systemic therapy. RET mutation status was detected in 90% of patients using NGS (89% tumor samples; 8% blood or plasma), and 10% using PCR. Of patients enrolled in LIBRETTO-531, 63% had M918T RET mutations and 37% had other RET mutations.
Efficacy results for LIBRETTO-531 based on the preplanned interim efficacy analysis are provided in Table 21 and Figure 2. At the time of this analysis, overall survival data were immature with 18 deaths observed (14% of pre-specified events).
|
Data from the pre-planned interim efficacy analysis. |
||
|
1 Based on the stratified Cox proportional hazard model. |
||
|
2 Based on stratified log-rank test. |
||
|
NR = Not reached; NE = not evaluable |
||
| RETEVMO N = 193 |
Cabozantinib or Vandetanib N = 98 |
|
| PFS | ||
| Number (%) of patients with an event | 26 (14%) | 33 (34%) |
| Median in months (95% CI) | NR (NE, NE) | 16.8 (12.2, 25.1) |
| Hazard ratio (95% CI)1 | 0.280 (95% CI: 0.165, 0.475) | |
| p-value2 | <0.0001 | |
| Overall Response Rate | ||
| ORR (95% CI) | 69% (62%, 76%) | 39% (29%, 49%) |
| Complete response | 12% | 4% |
| Partial response | 58% | 35% |
| Duration of Response | ||
| Median in months (95% CI) | NR (NE, NE) | 16.6 (10.4, NE) |
| Median follow-up time (months) | 11.1 | 12.8 |
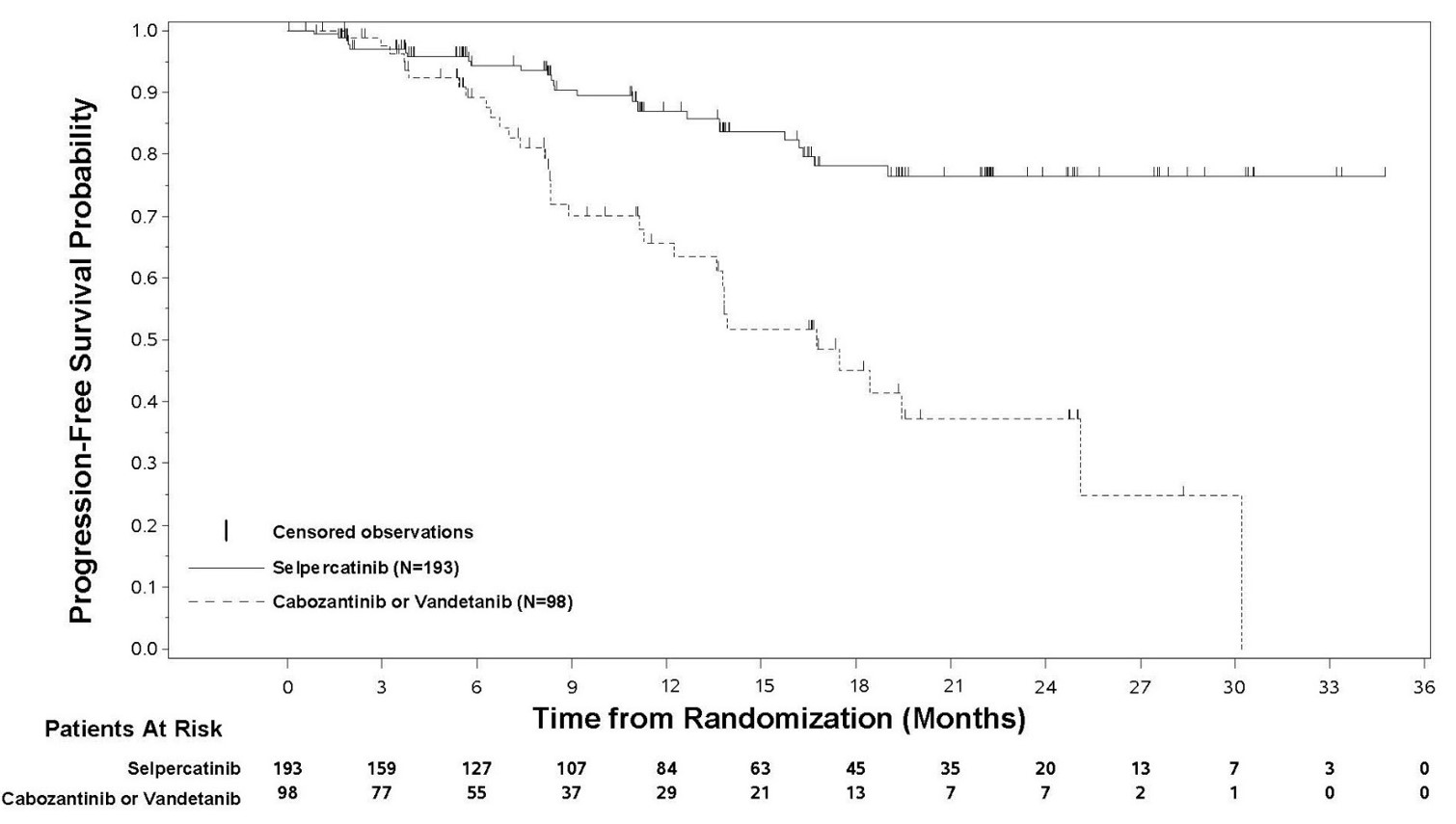
Figure 2: Kaplan-Meier Curves of Progression-Free Survival in LIBRETTO-531: RETEVMO versus Cabozantinib or Vandetanib
Patient-reported overall side effect impact was evaluated weekly in 222 patients (RETEVMO N = 145; cabozantinib or vandetanib N=77) who received at least one dose of treatment by at least 6 months prior to the data cutoff date and responded to the Functional Assessment of Cancer Therapy item GP5 (FACT GP5). Patient-reported overall side effect impact was derived as a proportion of time on treatment with high side effect bother (defined as response of 3 “Quite a bit” or 4 “Very much”) per FACT GP5.
Patient-reported overall side effect impact results for LIBRETTO-531 are provided in Table 22.
| RETEVMO (N=145) |
Cabozantinib or Vandetanib (N=77) |
|
| Mean proportion of time with high side effect bother (95% CI) (95% CI) | 8% (4.8%, 10%) | 24% (17%, 31%) |
| % Patients with high side effect bother 0% of time ≤25% of time |
61% 90% |
30% 66% |
Patient-reported overall side effect impact results were supported by a lower incidence of treatment discontinuation due to adverse reactions for RETEVMO (4.7%) compared to cabozantinib or vandetanib (27%) in patients who received at least one dose of study treatment. The median time on treatment at the data cutoff was 14.5 months in the RETEVMO arm and 8.3 months in the cabozantinib or vandetanib arm in patients who received at least one dose of study treatment.
LIBRETTO-121
The efficacy of RETEVMO was evaluated in pediatric and young adult patients with advanced RET-activated solid tumors enrolled in a multicenter, open-label, multi-cohort clinical trial (LIBRETTO-121, NCT03899792). Patients received RETEVMO 92 mg/m2 orally twice daily until disease progression, unacceptable toxicity, or other reason for treatment discontinuation. Tumor assessments were performed every 8 weeks for one year, then every 12 weeks; responses were assessed according to RECIST 1.1 per BIRC.
Efficacy was evaluated in 14 patients with RET-mutant MTC who were non-responsive to available therapies or had no standard systemic curative therapy available. The median age was 14 years (range 2 to 20); 64% were male; 71% were White, 14% were Black or African American; and 14% were Hispanic/Latino. Patients had metastatic (71%) or locally advanced (29%) disease; 43% had measurable disease at baseline; 21% had received prior systemic therapy. RET-mutant status was detected in 79% of patients using NGS tumor samples and in 21% using PCR.
Efficacy results for RET-mutant MTC in pediatric and young adult patients are summarized in Table 23.
|
1 Confirmed overall response rate assessed by BIRC. |
|
|
2 Based on observed duration of response. |
|
|
NR = not reached; NE = not estimable |
|
| RETEVMO (n = 14) |
|
| Overall Response Rate1 (95% CI) | 43% (18, 71) |
| Complete response | 7% |
| Partial response | 36% |
| Duration of Response | |
| Median in months (95% CI) | NR (NE, NE) |
| % with ≥12 months2 | 100% |
| % with ≥18 months2 | 67% |
14.3 RET Fusion-Positive Thyroid Cancer
LIBRETTO-001
The efficacy of RETEVMO was evaluated in patients with advanced RET fusion-positive thyroid cancer enrolled in a multicenter, open-label, multi-cohort clinical trial (LIBRETTO-001, NCT03157128). Efficacy was evaluated in 65 patients with RET fusion-positive thyroid cancer who were radioactive iodine (RAI)-refractory (if RAI was an appropriate treatment option) and were systemic therapy naïve and patients who were previously treated, in separate cohorts.
The median age was 59 years (range 20 to 88); 49% were male; 65% were White, 20% were Asian, 4.6% were Black or African American; and 11% were Hispanic/Latino. ECOG performance status was 0-1 (94%) or 2 (6%). All (100%) patients had metastatic disease with primary tumor histologies including papillary thyroid cancer (83%), poorly differentiated thyroid cancer (9%), anaplastic thyroid cancer (6%) and Hurthle cell thyroid cancer (1.5%). Previously treated patients had received a median of 1 prior therapy (range 1–4). RET fusion-positive status was detected in 97% of patients using NGS (89% tumor samples; 8% blood or plasma samples), and 3% using other local testing methods.
Efficacy results for RET fusion-positive thyroid cancer are summarized in Table 24.
|
1 Confirmed overall response rate assessed by BIRC. |
||
|
2 Based on observed duration of response. |
||
|
NE = not estimable |
||
| RETEVMO Previously Treated (n = 41) |
RETEVMO Systemic Therapy Naïve (n = 24) |
|
| Overall Response Rate1 (95% CI) | 85% (71%, 94%) | 96% (79%, 100%) |
| Complete response | 12% | 21% |
| Partial response | 73% | 75% |
| Duration of Response | ||
| Median in months (95% CI) | 26.7 (12.1, NE) | NE (42.8, NE) |
| % with ≥12 months2 | 54 | 65 |
Responses were observed in patients with each histology represented, including 3 of 4 patients with anaplastic thyroid cancer (all partial responses) and 6 of 6 patients with poorly differentiated thyroid cancer (1 complete response, 5 partial responses).
LIBRETTO-121
The efficacy of RETEVMO was evaluated in pediatric and young adult patients with advanced RET-activated solid tumors enrolled in a multicenter, open-label, multi-cohort clinical trial (LIBRETTO-121, NCT03899792) [see Clinical Studies (14.2)].
Efficacy was evaluated in 10 patients with RET fusion-positive thyroid cancer who were non-responsive to available therapies or had no standard systemic curative therapy available. The median age was 13.5 years (range 12 to 20); 60% were male; 40% were White, 50% were Asian; and 30% were Hispanic/Latino. All (100%) patients had metastatic disease and papillary thyroid cancer histology; 40% had measurable disease at baseline; 30% had received prior systemic therapy. RET fusion-positive status was detected in 90% of patients using NGS tumor samples and in 10% using FISH. Efficacy results for RET fusion-positive thyroid cancer in pediatric and young adult patients are summarized in Table 25.
|
1 Confirmed overall response rate assessed by BIRC. |
|
|
2 Based on observed duration of response. |
|
|
NR = not reached; NE = not estimable |
|
| RETEVMO (n = 10) |
|
| Overall Response Rate1 (95% CI) | 60% (26, 88) |
| Complete response | 30% |
| Partial response | 30% |
| Duration of Response | |
| Median in months (95% CI) | NR (NE, NE) |
| % with ≥12 months2 | 83% |
| % with ≥18 months2 | 50% |
14.4 Other RET Fusion-Positive Solid Tumors
LIBRETTO-001
The efficacy of RETEVMO was evaluated in patients with locally advanced or metastatic RET fusion-positive solid tumors enrolled in a multicenter, open-label, multi-cohort clinical trial (LIBRETTO-001, NCT03157128). Efficacy was evaluated in 41 patients with RET fusion-positive tumors other than NSCLC and thyroid cancer with disease progression on or following prior systemic treatment or who had no satisfactory alternative treatment options.
The median age was 50 years (range 21 to 85), 54% were female, 68% were White, 24% were Asian, and 4.9% were Black; and 7% were Hispanic/Latino. ECOG performance status was 0-1 (95%) or 2 (5%) and 95% of patients had metastatic disease. Thirty-seven patients (90%) received prior systemic therapy (median 2 [range 0 – 9]; 32% received 3 or more). The most common cancers were pancreatic adenocarcinoma (27%), colorectal (24%), salivary (10%) and unknown primary (7%). RET fusion-positive status was detected in 97.6% of patients using NGS and 2.4% using FISH.
Efficacy results for RET fusion-positive solid tumors other than NSCLC and thyroid cancer are summarized in Table 26 and Table 27.
|
1 Confirmed overall response rate assessed by BIRC. |
|
|
2 Based on observed duration of response. |
|
|
NE = not estimable |
|
| RETEVMO (n = 41) |
|
| Overall Response Rate1 (95% CI) | 44% (28, 60) |
| Complete response | 4.9% |
| Partial response | 39% |
| Duration of Response | |
| Median in months (95% CI) | 24.5 (9.2, NE) |
| % with ≥6 months2 | 67% |
|
+ denotes ongoing response. |
||||
|
1 Confirmed overall response rate assessed by BIRC. |
||||
|
2 Best overall response for each patient is presented for tumor types with ≤2 patients. |
||||
|
CI = confidence interval, CR = complete response, DOR = duration of response, NA = not applicable, NE = not evaluable, ORR = overall response rate, PR = partial response, SD = stable disease. |
||||
| Tumor Type | Patients (n = 41) |
ORR1,2 | DOR Range (months) |
|
| n (%) | 95% CI | |||
| Pancreatic adenocarcinoma | 11 | 6 (55%) | (23, 83) | 2.5, 38.3+ |
| Colorectal | 10 | 2 (20%) | (2.5, 56) | 5.6, 13.3 |
| Salivary | 4 | 2 (50%) | (7, 93) | 5.7, 28.8+ |
| Unknown primary | 3 | 1 (33%) | (0.8, 91) | 9.2 |
| Breast | 2 | PR, CR | NA | 2.3+, 17.3 |
| Sarcoma (soft tissue) | 2 | PR, SD | NA | 14.9+ |
| Xanthogranuloma | 2 | NE, NE | NA | NA |
| Carcinoid (bronchial) | 1 | PR | NA | 24.1+ |
| Carcinoma of the skin | 1 | NE | NA | NA |
| Cholangiocarcinoma | 1 | PR | NA | 5.6+ |
| Ovarian | 1 | PR | NA | 14.5+ |
| Pulmonary carcinosarcoma | 1 | NE | NA | NA |
| Rectal neuroendocrine | 1 | NE | NA | NA |
| Small intestine | 1 | CR | NA | 24.5 |
LIBRETTO-121
The efficacy of RETEVMO was evaluated in pediatric and young adult patients with advanced RET-activated solid tumors enrolled in a multicenter, open-label, multi-cohort clinical trial (LIBRETTO-121, NCT03899792) [see Clinical Studies (14.2)].
Efficacy was evaluated in one patient with locally advanced refractory RET-fusion positive malignant peripheral nerve sheath tumor who did not respond. Responses were observed in patients with RET fusion-positive thyroid cancer [see Clinical Studies (14.3)].
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
RETEVMO capsules are supplied as follows:
| Capsule Strength | Description | Package Configuration | NDC Number |
| 40 mg | Gray opaque, imprinted with “Lilly”, “3977” and “40 mg” in black ink | 60 count bottle | NDC 0002-3977-60 |
| 80 mg | Blue opaque, imprinted with “Lilly”, “2980” and “80 mg” in black ink | 60 count bottle | NDC 0002-2980-60 |
| 120 count bottle | NDC 0002-2980-26 |
RETEVMO tablets are supplied in bottles with desiccant in the following configurations:
| Tablet Strength | Description | Package Configuration | NDC Number |
| 40 mg | Light gray, film coated, round tablets debossed with “Ret 40” on one side and “5340” on the other side | 60 count bottle | NDC 0002-5340-60 |
| 80 mg | Dark red-purple, film coated, round tablets debossed with “Ret 80” on one side and ”6082” on the other side | 60 count bottle | NDC 0002-6082-60 |
| 120 mg | Light purple, film coated, round tablets debossed with “Ret 120” on one side and “6120” on the other side | 60 count bottle | NDC 0002-6120-60 |
| 160 mg | Light pink, film coated, round tablets debossed with Ret “160” on one side and “5562” on the other side | 60 count bottle | NDC 0002-5562-60 |
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Hepatotoxicity
Advise patients that hepatotoxicity can occur and to immediately contact their healthcare provider for signs or symptoms of hepatotoxicity [see Warnings and Precautions (5.1)].
Interstitial Lung Disease (ILD)/Pneumonitis
Advise patients that ILD/ pneumonitis can occur and to contact their healthcare provider immediately for signs or symptoms of ILD including new or worsening cough or shortness of breath [see Warnings and Precautions (5.2)].
Hypertension
Advise patients that they will require regular blood pressure monitoring and to contact their healthcare provider if they experience symptoms of increased blood pressure or elevated readings [see Warnings and Precautions (5.3)].
QT Prolongation
Advise patients that RETEVMO can cause QTc interval prolongation and to inform their healthcare provider if they have any QTc interval prolongation symptoms, such as syncope [see Warnings and Precautions (5.4)].
Hemorrhagic Events
Advise patients that RETEVMO may increase the risk for bleeding and to contact their healthcare provider if they experience any signs or symptoms of bleeding [see Warnings and Precautions (5.5)].
Hypersensitivity Reactions
Advise patients to monitor for signs and symptoms of hypersensitivity reactions, particularly during the first month of treatment [see Warnings and Precautions (5.6)].
Tumor Lysis Syndrome
Advise patients to contact their healthcare provider promptly to report any signs and symptoms of TLS [see Warnings and Precautions (5.7)].
Risk of Impaired Wound Healing
Advise patients that RETEVMO may impair wound healing. Advise patients to inform their healthcare provider of any planned surgical procedure [see Warnings and Precautions (5.8)].
Hypothyroidism
Advise patients that RETEVMO can cause hypothyroidism and to immediately contact their healthcare provider for signs or symptoms of hypothyroidism [see Warnings and Precautions (5.9)].
Slipped Capital Femoral Epiphysis/Slipped Upper Femoral Epiphysis
Advise pediatric patients and caregivers to contact their healthcare provider promptly to report any signs and symptoms indicative of slipped capital femoral epiphysis/slipped upper femoral epiphysis [see Warnings and Precautions (5.11)].
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential of the possible risk to a fetus. Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.10), Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective contraception during the treatment with RETEVMO and for 1 week after the last dose [see Use in Specific Populations (8.3)].
Advise males with female partners of reproductive potential to use effective contraception during treatment with RETEVMO and for 1 week after the last dose [see Use in Specific Populations (8.3)].
Lactation
Advise women not to breastfeed during treatment with RETEVMO and for 1 week after the last dose [see Use in Specific Populations (8.2)].
Infertility
Advise males and females of reproductive potential that RETEVMO may impair fertility [see Use in Specific Populations (8.4), Nonclinical Toxicology (13.1)].
Drug Interactions
Advise patients and caregivers to inform their healthcare provider of all concomitant medications, including prescription medicines, over-the-counter drugs, vitamins, and herbal products. Inform patients to avoid St. John's wort, proton pump inhibitors, H2 receptor antagonists, and antacids while taking RETEVMO.
If PPIs are required, instruct patients to take RETEVMO with food. If H2 receptor antagonists are required, instruct patients to take RETEVMO 2 hours before or 10 hours after the H2 receptor antagonist. If locally-acting antacids are required, instruct patients to take RETEVMO 2 hours before or 2 hours after the locally-acting antacid [see Drug Interactions (7.1, 7.2)].
Marketed by: Lilly USA, LLC, Indianapolis, IN 46285, USA
Copyright © 2020, 2024, Eli Lilly and Company. All rights reserved.
RET-0007-USPI-20240927
|
This Patient Information has been approved by the U.S. Food and Drug Administration. |
09/2024 | ||||||||||||||||
| PATIENT INFORMATION | |||||||||||||||||
| RETEVMO®(reh-TEHV-moh) (selpercatinib) capsules |
RETEVMO®(reh-TEHV-moh) (selpercatinib) tablets |
||||||||||||||||
| What is RETEVMO? RETEVMO is a prescription medicine that is used to treat certain cancers caused by abnormal RET genes in:
|
|||||||||||||||||
| Your healthcare provider will perform a test to make sure that RETEVMO is right for
you. It is not known if RETEVMO is safe and effective when used:
|
|||||||||||||||||
Before taking RETEVMO, tell your healthcare provider about all your medical conditions,
including if you:
|
|||||||||||||||||
| Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
RETEVMO may affect the way other medicines work, and other medicines may affect how
RETEVMO works, and may increase your risk of side effects. During treatment with RETEVMO, you should avoid taking:
|
|||||||||||||||||
| If you cannot avoid taking PPIs, H2 blockers, or antacids, see “How should I take RETEVMO?” for more information on how to take RETEVMO with these medicines. Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine. |
|||||||||||||||||
|
|||||||||||||||||
|
What are the possible side effects of RETEVMO?
|
|||||||||||||||||
| The most common side effects of RETEVMO in adults with solid tumors include: | |||||||||||||||||
|
|
||||||||||||||||
| The most common side effects of RETEVMO in children 2 years and older with solid tumors include: | |||||||||||||||||
|
|
||||||||||||||||
| The most common severe abnormal laboratory test results with RETEVMO in adults with
solid tumors include decreased white blood cell count, increased liver enzymes, decreased levels of sodium
in the blood, and decreased levels of calcium in the blood. The most common severe abnormal laboratory test results with RETEVMO in children 2 years and older with solid tumors include decreased levels of calcium in the blood, decreased red blood cell count, and decreased white blood cell count. RETEVMO may affect fertility in females and males, which may affect your ability to have children. Talk to your healthcare provider if this is a concern for you. These are not all the possible side effects with RETEVMO. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|||||||||||||||||
How should I store RETEVMO?
|
|||||||||||||||||
| Keep RETEVMO and all medicines out of the reach of children. | |||||||||||||||||
| General information about the safe and effective use of RETEVMO. Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use RETEVMO for a condition for which it was not prescribed. Do not give RETEVMO to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for more information about RETEVMO that is written for health professionals. |
|||||||||||||||||
| What are the ingredients in RETEVMO? Active ingredient: selpercatinib Capsules: colloidal silicon dioxide and microcrystalline cellulose. The 40 mg capsule shell contains: gelatin, titanium dioxide, ferric oxide black and black ink. The 80 mg capsule shell contains: gelatin, titanium dioxide, FD&C blue #1 and black ink. The black ink contains: shellac, potassium hydroxide and ferric oxide black. Tablets: croscarmellose sodium, hydroxypropyl cellulose, mannitol, microcrystalline cellulose, and sodium stearyl fumarate. The tablet film coating material contains polyvinyl alcohol, titanium dioxide, polyethylene glycol, and talc. Additionally, the film coating of the 40 mg, 80 mg, and 120 mg tablets contains ferrosoferric oxide and the film coating of the 80 mg, 120 mg, and 160 mg tablets contain ferric oxide. Marketed by: Lilly USA, LLC, Indianapolis, IN 46285, USA Copyright © 2020, 2024, Eli Lilly and Company. All rights reserved. RET-0007-PPI-20240927 |
|||||||||||||||||
PACKAGE LABEL - Retevmo 40 mg 60 Count Bottle
NDC-0002-3977-60
60 capsules
Rx only
Retevmo™
(selpercatinib)
capsules
40 mg
Each capsule contains
40 mg selpercatinib
www.retevmo.com
Lilly

PACKAGE LABEL - Retevmo 80 mg 60 Count Bottle
NDC-0002-2980-60
60 capsules
Rx only
Retevmo™
(selpercatinib)
capsules
80 mg
Each capsule contains
80 mg selpercatinib
www.retevmo.com
Lilly

PACKAGE LABEL – Retevmo 40mg Tablets
NDC 0002-5340-60
60 tablets Rx only
Retevmo®
(selpercatinib)
tablets
40 mg
Each tablet contains 40 mg selpercatinib
www.retevmo.com
Lilly

PACKAGE LABEL – Retevmo 80mg Tablets
NDC 0002-6082-60
60 tablets Rx only
Retevmo®
(selpercatinib)
tablets
80 mg
Each tablet contains 80 mg selpercatinib
www.retevmo.com
Lilly


